항수초희소돌기아교세포당단백질항체 관련 질환
Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: A Review
Article information
Trans Abstract
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is an inflammatory demyelinating disease of the central nervous system that is distinct from multiple sclerosis and neuromyelitis optica spectrum disorder. While an international consensus has recently been established regarding the clinical features and diagnostic criteria of MOGAD, further studies are needed to improve our understanding of the disease, including its treatment and prognosis. Accurate diagnostic methods and a comprehensive understanding of the clinical features are crucial, as test results must be integrated with clinical considerations. Currently, there is a lack of randomized controlled clinical trials on the treatment of MOGAD, but observational studies suggest that immunotherapy may be effective. This review aims to provide a comprehensive overview of the pathophysiology, clinical features, and recently established diagnostic criteria of MOGAD, as well as available treatment options, and prognosis.
서론
Myelin oligodendrocyte glycoprotein (MOG)은 중추신경계의 수초에 발현하는 당단백이다. 예전부터 항MOG항체(anti-MOG antibody, MOG항체)의 존재는 알려져 왔으나, 혈청에서 생세포기반검사(live cell-based assay)를 통한 MOG항체의 검출법이 알려지면서부터 MOG항체의 임상적 중요성이 대두되었고, 이로 인해 중추신경계 염증 탈수초질환 중에서 MOG 항체질환(MOG antibody-associated disease, MOGAD)이라는 독립된 질환으로의 분류가 가능하게 되었다.1,2
MOGAD는 다발성경화증(multiple sclerosis, MS)이나 항아쿠아포린4항체 양성 시신경척수염범주질환(anti-aquaporin 4 antibody positive neuromyelitis optica spectrum disorder, AQP4+NMOSD)과는 중추신경계 염증 탈수초질환이라는 공통점이 있지만, 임상 경과, 병리학적 특징, 병태생리, 영상의학적 특징, 치료 반응, 예후 등 많은 부분에서 차이점이 있다.2-6 모든 연령대에서 발생할 수 있으며, 전 세계적으로 유병률은 10만 명당 2.0명, 발병률은 연간 10만 명당 0.2-0.3명으로 추정된다.7,8 성인 중추신경계 염증 탈수초질환 중에는 약 1-6%를 차지하고, 소아에서는 급성 탈수초질환의 약 40% 정도까지 보고된다. 유병률은 점차 증가하고 있고, 최근 MOGAD의 국제 진단 기준3이 제시됨에 따라 향후 더 유동적일 것으로 예상된다.
본 종설에서는 MOGAD의 병태생리, 임상 양상, 임상 경과, 검사법과 진단 기준 및 치료와 예후에 대한 전반적인 내용을 살펴보고자 한다.
본문
MOGAD의 병태생리
MOG항체의 생성을 유발하는 인자는 명확히 알려지지 않았지만 말초면역계(peripheral immune system)에서 자가면역 기전에 의해 생긴다고 여겨지고 있다.9 MOG항체 생성에는 B세포나 형질세포(plasma cell) 외에도, B세포가 형질세포로 분화하도록 도와주는 항체특이적 여포보조T세포(antigen-specific T follicular helper cell)가 연관되어 있다고 추측된다. MOG항체가 병을 일으키기 위해서는 중추신경계 내로 유입되어야 한다. 혈액뇌장벽(blood-brain barrier, BBB)이 손상되어 활성화된 B세포, 형질세포 및 MOG항체가 BBB를 통과하면, 중추신경계 내에서는 MOG항체가 MOG를 발현하는 수초에 결합하여 수초손상 및 탈수초를 진행시킨다. 이때, 대식세포(macrophage)와 T세포(주로 CD4+ T세포), 과립구(granulocyte) 등이 같이 중추신경계 내로 유입되며, 사실 병변에는 이들 세포가 B세포보다 더 많은 비율을 차지하고 있어 보체 활성을 일으키기도 한다.10,11 이로 인해 MOGAD의 병리학적 소견은 정맥주위 합류성 탈수초 병변, 피질내 탈수초 병변, 병변 내 MOG 발현 수초 소실, CD4+ T세포 및 과립구 우세 염증, 활동성 백질 병변 내 보체침착, 부분적 축삭 보존 등으로 특징지을 수 있다. MS와는 달리 CD8+ T세포보다 CD4+ T세포가 우세하고, 서서히 확장되는 플라크(slowly expending plaque)가 없으며, AQP4+NMOSD와는 달리 AQP4가 잘 보존되어있고, AQP4 주변의 보체 침착이 관찰되지 않는다.5,6 또한 전염증성 사이토카인(proinflammatory cytokine) 및 B세포 사이토카인/케모카인이 증가됨이 뇌척수액 또는 혈액에서 확인되었다.12,13
MOGAD의 임상 양상
MOGAD는 MS나 AQP4+NMOSD와 같이 여성에서 우세하지는 않고, 전 연령대에서 발생할 수 있으나 성인과 소아에서 발현하는 임상 양상 표현형에 차이가 있다(Table 1).14,15 성인에서는 시신경염(optic neuritis)이, 소아에서는 급성파종뇌척수염(acute disseminated encephalomyelitis, ADEM)이 가장 흔한 임상이다. 특히 소아에서는 전체 ADEM 환자 중 많게는 64%까지, 그리고 재발 경과를 보이는 ADEM 환자는 대부분 MOGAD로 진단된다.16,17 그 외에 척수염 단독 또는 시신경염과 척수염이 동반되는 경우 또한 흔하게 관찰되며, 특히 AQP4 항체 음성인 NMOSD 또는 종단광범위횡단척수염(longitudinally extensive transverse myelitis, LETM)의 상당수에서 MOGAD로 진단되기도 한다.18,19 그 외에 일부 환자들은 대뇌피질뇌염, 뇌간 및 소뇌 병변, 종괴형성 탈수초 병변, 뇌신경병증, 진행성 백질 손상 등으로 발현한다. 침범 부위에 따른 임상적 특징은 다음과 같다.
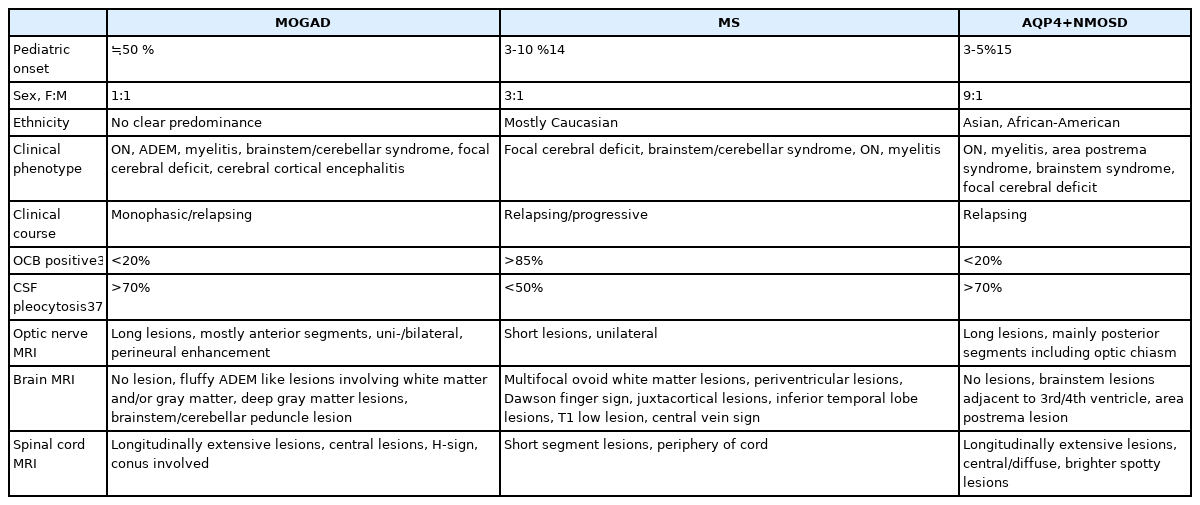
Clinical and radiological features of MOGAD, MS, and AQP4+NMOSD
시신경 병변
MOGAD 환자의 약 80%까지 시신경이 침범될 수 있으며, 특히 양측성(31-58%), 재발성으로 발생할 수 있다.20,21 대표적인 증상/증후로는 중심시력저하, 안구뒤통증, 색각이상, 구심동공결손(afferent pupillary defect) 등이 있고, 검진에서는 시신경유두 부종이 흔히 관찰된다. 신경생리검사에서는 시각유발전위(visual evoked potential), 광간섭단층촬영(optical coherence tomography, OCT) 등의 검사에 이상을 보일 수 있으나 진단에 특이적이지는 않다. 시신경염 단독으로도 나타나지만, ADEM이나 척수염과 동반되어 나타나기도 한다.
시력저하는 환자마다 차이가 있지만 종종 매우 심하게 나타나기도 한다. 다만 스테로이드 치료에 반응이 좋아 빠르게 회복될 수 있으나, 스테로이드를 줄이거나 중단한 뒤 바로 재발할 수도 있다.22,23 약 30-50%의 MOG 항체 양성 시신경염은 재발하는 경과를 보이는데, 처음에는 ADEM이나 NMOSD에 동반하여 발병하였다가 시신경염으로 지속적으로 재발하기도 한다.24
진단을 위한 안구 MRI 촬영 시에는 T2강조영상 및 T1조영증강영상에서 고신호강도를 보이며, 특히 지방억제(fat-suppression)기법을 이용한 조영증강영상을 촬영하는 것이 시신경 염증 판단에 유리하다(Fig. 1A). MS 또는 AQP4+NMOSD와의 차이점으로는 시신경 중 안와(orbital) 부위를 자주 침범하여 시신경의 머리부분 부종이 나타날 수 있고, 이로 인한 안구뒤통증이 더 호발하기도 한다. 시신경 병변은 길게 침범하는 것이 특징적이며, 시신경집(optic nerve sheath)을 침범하여 조영증강을 나타낼 수 있다.18,25 또한, 시신경유두부종 및 출혈이 더 흔하게 관찰될 수 있어 이를 확인할 수 있는 안저검사가 유용하다. 시신경염 급성기 때 OCT로 시신경유두주위망막신경섬유층(peripapillary retinal nerve fiber layer)이 두꺼워진 것을 확인할 수 있고, 이는 급성기가 지나면 점차 얇아지는데, 특히 측두사분면(temporal quadrant)이 더 심하게 얇아진다. 시신경염에서는 황반부 신경절세포-내망상층(ganglion cell-inner plexiform layer)의 두께 또한 얇아질 수 있는데, MOGAD와 관련된 시신경염은 AQP4+NMOSD나 MS와 비교해서 그 정도가 경미하여 망막의 손상이 덜하다고 유추할 수 있다.26
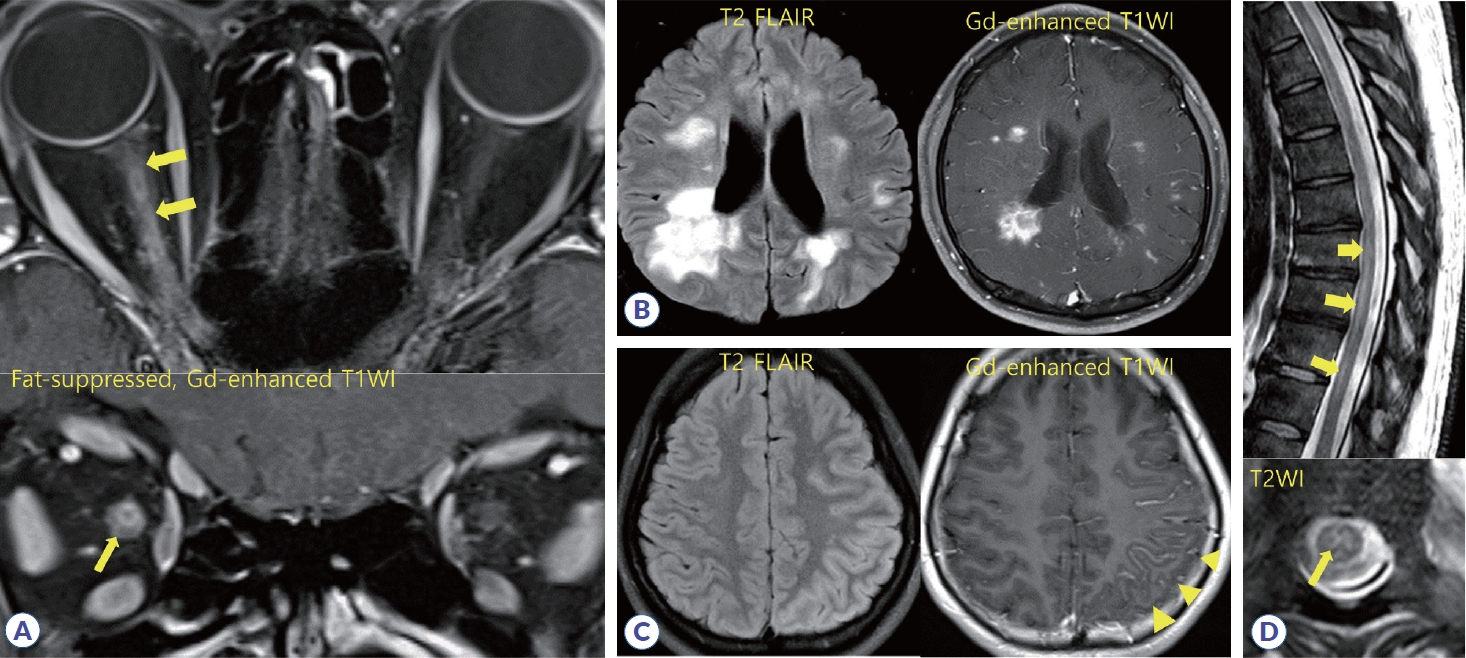
Common radiological features of MOGAD. (A) High signal intensity is seen along the right optic nerve (top), and perineural enhancement (bottom) is visible (yellow arrows) on fat-suppressed, gadolinium-enhanced T1-weighted image compared to the normal left optic nerve. (B) Multiple confluent white matter lesions with gadolinium enhancement in a patient present as acute disseminated encephalomyelitis. (C) Fluid attenuated inversion recovery, hyperintense cortical lesions, and prominent leptomeningeal enhancement (arrowheads) are visible along with cerebral cortex. (D) Longitudinally extensive transverse myelitis is common and may show the H-sign due to involvement of gray matter (yellow arrows). Gd, gadolinium; WI, weighted image; FLAIR, fluid attenuated inversion recovery; MOGAD, myelin oligodendrocyte glycoprotein antibody-associated disease.
뇌 및 뇌간 병변
MOGAD 환자에서 뇌/뇌간을 침범하는 환자는 ADEM, 대뇌피질뇌염, 뇌간 및 소뇌증후군 등의 양상으로 발현한다. 무증상 뇌병변도 있지만, 시신경염이나 척수염으로 처음 발현한 환자에서 뇌 병변이 없는 경우가 약 47-68%로 더 많아 MS와 구별될 수 있다.4 뇌병변은 양측성, 경계가 불명확한(ill-defined), 큰 병변이 많고(Fig. 1B), 간혹 심부회색질(deep gray matter)을 침범하기도 한다. 전형적인 MS 병변이나 지속적인 T1 저강도신호 병변은 MOGAD에서는 드물다. 뇌간 병변의 경우 교뇌침범이 흔하며, 특히 중간소뇌다리를 침범하는 큰 병변의 경우에는 MS나 AQP4+NMOSD보다 MOGAD를 시사하고,27 다만 맨아래구역증후군(area postrema syndrome)으로 나타나는 경우는 매우 드물다.28 소아에서는 MOGAD 환자의 약 50%를 차지할 정도로 ADEM이 가장 흔하지만 성인에서는 약 5.6%로 많지 않다.19 특히 소아 MOGAD는 ADEM 전에 감염성 질환이 선행하는 경우가 40-75%로 많고, 약 70% 환아는 영상의학적으로 관해가 된다.29 하지만 백질형성장애 유사 병변(leukodystrophy like lesion) 양상으로 나타나는 경우는 예후가 불량하고, MOG항체가 양성인 ADEM의 경우 음성인 경우보다 재발을 잘 한다. 대뇌 뇌염은 발열, 두통, 의식저하, 발작 등의 증상을 보이며, 이 중 발작과 함께 대뇌 피질에 액체감쇠역전회복영상(fluid attenuated inversion recovery, FLAIR) MRI에서 고신호강도를 보이는 병변이 있는 경우를 FLAMES (FLAIR hyperintense lesions in anti-MOG encephalitis with seizures)로 명칭하였다(Fig. 1C).30 그 외에, NMDA수용체 항체와 MOG항체가 동시에 양성인 뇌염도 있다.31,32
척수 병변
척수 병변은 척수염 단독 또는 ADEM이나 시신경염과 동반하여 나타날 수 있다. 감각이상, 근위약, 괄약근장애, 성기능장애 등이 발생할 수 있으며, 증상 중증도는 50% 이상의 환자가 확장장애상태척도(Expanded Disability Status Scale, EDSS) 4점을 초과하는 중등도 또는 심한 증상으로 나타나지만 스테로이드 치료에 대부분 운동기능 회복에는 반응이 좋다. 배뇨 및 배변장애나 성기능장애는 지속적으로 나타날 수 있고, 통증성 긴장연축(painful tonic spasm)은 AQP4+NMOSD만큼 흔하지는 않다.33,34
MRI 촬영 시 T2강조영상에서 고신호강도를 보이는 병변이 보이나, 약 10% 정도는 첫 증상 발생 시기에 MRI가 정상일 수 있다.35 병변은 단독 또는 다수의 LETM으로 나타나는 경우가 60% 정도로 많고, 약 30-50%에서는 회색질을 침범하여 H-sign이 보이거나 척수원뿔을 침범하는 경우도 약 26% 정도로 MS나 AQP4+NMOSD와 비교하여 특징적이다(Fig. 1D).33 대부분 척수의 T2고신호강도는 시간이 지나면 줄어들거나 없어지기도 하지만, 심한 척수병인 경우 척수 위축이 나타나는 경우도 있다.34,36 시신경이나 뇌병변 증상을 보이는 환자에서도 무증상 척수 병변이 발견되기도 하며, 척수병 증상으로 발현하더라도 뇌 또는 시신경에 무증상 병변이 발견되는 경우도 약 33-50% 정도 된다.
MOGAD의 진단
MOG항체검사
MOG항체검사는 혈청에서 시행하는 것을 우선으로 하며, 경우에 따라서는 뇌척수액에서 시행할 수 있다.1,3 혈청 MOG항체 검사는 인간 MOG의 전체 길이의 단백질을 이용한 생세포기반 분석(live cell-based assay)을 통해 MOG-immunoglobulin G (MOG-IgG)를 검출하는 것을 권장한다. MOG-IgG가 IgG1이기 때문에, 검사에 쓰이는 2차항체는 IgG Fc나 IgG1를 사용하되, 검증된 검사실일 경우 IgG(H+L)를 사용할 수 있다.39 고정세포기반분석(fixed cell-based assay)은 생세포기반분석이 불가능할 경우 대체 검사법으로 사용할 수 있지만, 생세포기반분석법보다 민감도와 특이도가 낮으니 주의해야 한다. 효소결합면역 흡착측정법(enzyme-linked immunosorbent assay)은 민감도와 특이도가 낮아 MOG-IgG 측정에는 권장되지 않는다.40
‘MOG항체 명확한 양성’의 기준은 생세포기반분석일 경우 양성기준역가보다 최소 2번 더 희석하여도 양성일 때나 분석법에서 정한 특정 역가를 넘을 때, 또는 형광활성세포선별기(fluorescence-activated cell sorting)를 이용한 유세포분석법(flow cytometry) 기준비(cut-off ratio)를 넘을 때로 하며, 고정세포기반분석일 경우는 역가가 1:100 이상일 때로 한다. ‘MOG 항체 약양성’은 생세포기반분석은 각 분석에서 정한 역가를 기준보다 낮을 때, 고정세포기반분석은 1:10 이상이고 1:100 미만일 때로 한다. MOG항체의 역가가 높은 경우 실험실 간 재현성이 좋지만, 낮은 경우에는 실험실 간 양성 판단에 재현성이 떨어진다. 또한 높은 역가를 적용할수록 MOG항체검사의 양성예측도가 높아진다.40,41
단독으로 뇌척수액에서 MOG항체를 검사하면 혈청 MOG항체검사보다 민감도가 떨어지지만, 일부 탈수초질환 환자에서 혈청 MOG항체가 음성이더라도 뇌척수액에서는 MOG항체가 양성이었고, 이들이 MOGAD 임상 양상에 부합하였기 때문에, 임상적, 영상의학적으로 MOGAD가 의심되는 경우 혈청 MOG항체가 음성이라면, 뇌척수액 MOG항체검사가 도움이 될 수 있다.42-44
검사 시점은 임상 증상이 나타났을 때, 특히 급성기 치료 전에 검사할 경우 가장 MOG항체가 검출될 가능성이 높다. 만약 급성기 치료가 시작된 이후 검사가 시행되었고, MOG항체가 음성인 경우, MOGAD가 의심된다면, 급성기 치료 효과 종료 후 최소 3개월이 지난 다음이나, 재발하였을 경우 다시 검사를 시행할 것이 권장된다.1,45
MOGAD 진단 기준
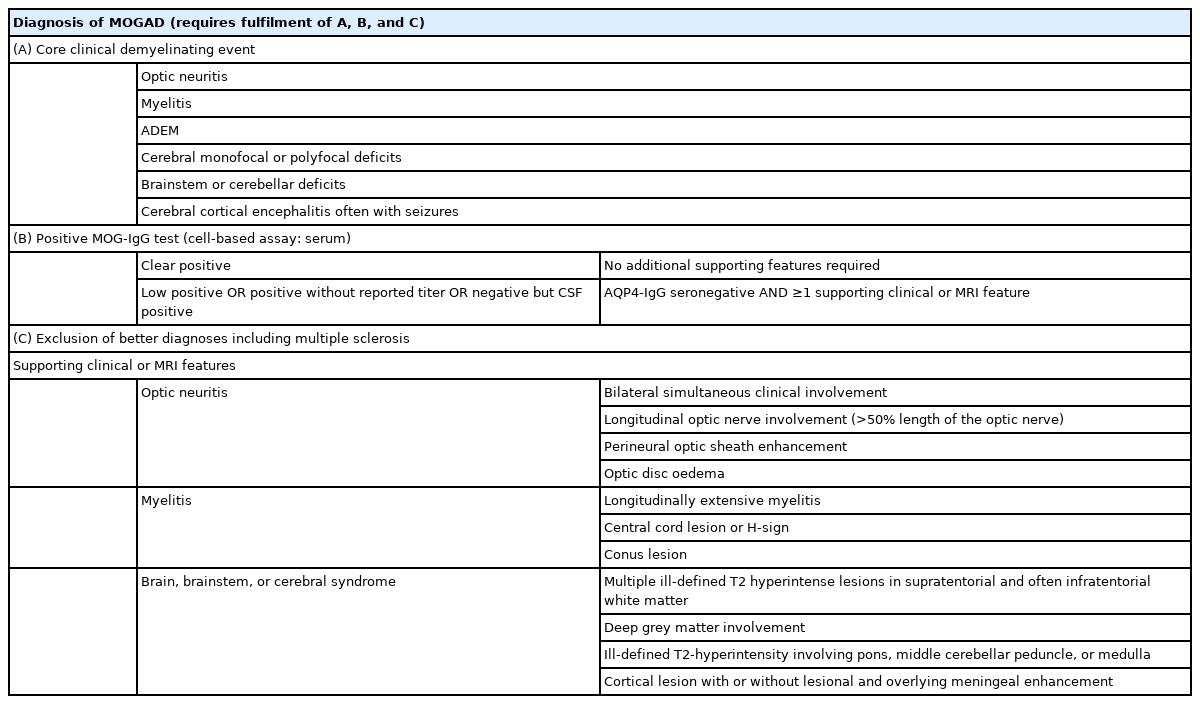
2023년 처음으로 MOGAD의 진단 기준이 제안되었다.3 MOGAD의 진단을 위해서는 임상 양상과 증상 발생을 확인하기 위한 신경학적 검진과 영상의학적 소견 및 검사실 소견을 만족해야 한다. 특히 모든 탈수초질환에서 스크리닝 목적으로 MOG항체검사를 하는 것은 지양해야 한다.3
제시된 진단 기준은 Table 2와 같다. 핵심임상증상유형(core clinical attack type)과 혈청 ‘MOG항체 명확한 양성’인 경우, MOGAD로 진단할 수 있다. 혈청 ‘MOG항체 약양성’인 경우, 역가가 제시되지 않은 고정세포기반분석 양성인 경우, 또는 혈청 음성이지만 뇌척수액 ‘MOG항체 명확한 양성’인 경우에는 핵심임상증상유형이 있더라도 보조임상/MRI 양상(supporting clinical or MRI features) 중 한 개 이상이 MOGAD로 진단할 수 있다. 모든 진단에 앞서 더 확실한 다른 진단이 있다면 배제되어야 한다. 특히 MS의 경우 0.3-2.5%에서 MOG항체 양성이 나타날 수 있기 때문에 이들이 MOGAD로 오진되지 않도록 주의해야 한다. 다만, 11세 미만의 소아에서는 오히려 중추신경계 탈수초질환 중에는 MS가 드물고 MOG항체 양성인 환아가 많기 때문에 MOG항체검사가 필요할 수 있다. 항아쿠아포린4항체와 MOG항체가 둘 다 양성인 경우는 매우 드물게 보고되고 있다. 하지만 ‘MOG항체 약양성’이고 항아쿠아포린4항체가 양성이라면 진단 기준에서도 제시된 바와 같이 AQP4+NMOSD 가능성이 더 높다. 감별에 주의해야하는 red-flag 증후로는 ① 임상적 재발 없이 신경학적 증상의 지속적 진행하는 경우, ② 증상의 가장 심한 정점까지 수분에서 수시간 안에 초급성으로 발병하는 경우, ③ 급성기 시 고용량 스테로이드 치료에 호전이 없는 경우, ④ 다발성경화증에 전형적인 MRI 소견과 함께 올리고클론 띠 양성인 경우, ⑤ 무증상 T2 고신호강도 병변이 발생하거나 기존 병변이 대부분 그대로 유지되는 경우, ⑥ 6개월 이상 지속되는 조영증강 병변이 있는 경우 등이 있다.3
MOGAD의 치료
MOGAD의 치료는 다른 중추신경계 염증 탈수초질환과 마찬가지로 급성기 치료와 장기적 재발 방지 치료로 나뉠 수 있다. 여기서 재발의 정의는 첫 증상이 발생한지 30일이 경과한 후 새로운 증상이 다시 나타난 것으로 하였다.3
급성기 치료
현재까지는 MOGAD 급성기 치료에 대한 무작위 대조 시험(randomized controlled trial)이나 근거중심(evidence-based) 지침이 없다. 또한 MOG항체 양성 여부가 중추신경계 탈수초질환의 급성기 치료에 영향을 준다는 근거가 없기 때문에, 대부분 중추신경계 염증 탈수초질환의 급성기 치료에 따르고 있다. 특히, MOG항체검사 결과 확인이 급성기 치료를 지연시켜서는 안된다.
급성기 치료 1차 약제는 경정맥 메틸프레드니솔론으로, 30 mg/kg/day 또는 1 g/day 용량으로 3-5일 연속으로 치료한다. MOGAD는 여러 관찰 연구에서 경정맥 고용량 스테로이드 치료 반응이 좋은 것으로 알려져 있다.23,46 치료 효과가 충분치 않은 경우, 혈장분리교환술(plasmapheresis), 면역글로불린 정맥 주사(intravenous immunoglobulin, IVIG), 또는 혈장분리교환술 후 IVIG를 고려할 수 있다.47 혈장분리교환술은 통상적으로는 매번 혈장 부피의 1-1.5배 정도를 교체하며, 격일로 총 5-7번 시행한다. 드물게 중심정맥관 삽입과 관련된 출혈이나 감염 위험, 시트르산에 의한 저칼슘혈증, 대사산증 등이 발생할 수 있다. 특히, 혈장 아닌 알부민 등으로 교체할 경우 응고인자가 결핍될 수 있으므로 주의해야 한다. IVIG는 일반적으로 0.4 g/kg/day 용량을 5일 동안 투여하여 총 2 g/kg을 투여한다. 부작용은 드물게 두통, 무력감, 열, 발진, 혈액응고이상, 신장기능저하, 아나필락시스 등이 발생할 수 있다.
고용량 경정맥 스테로이드 치료 후에 경구용 스테로이드 치료 여부, 기간, 용량은 증상의 중증도, 재악화 위험성을 고려하여 결정하는데, MOGAD는 스테로이드 치료에 의존성이 높아 감량 또는 중단 시에 재발할 위험성이 있고, 재발 또한 2개월 이내에 많이 발생하므로, 최소 3개월 이상에 걸쳐 서서히 감량하는 것이 권장된다.24
장기적 재발 방지 치료
중추신경계 염증 탈수초질환에서 재발성 경과를 보이는 질환의 경우 장기적인 재발 방지 치료가 필요하지만, MOGAD는 단발성 경과와 재발성 경과가 모두 가능한 질환이다. 아직은 MOGAD 환자에서 재발할 위험성을 평가하거나 장기적 예후를 예측할 수 있는 명확한 예측인자는 없다. 특히 소아에서는 약 70% 정도의 환자가 단발성 증상에 그치기 때문에, 장기적인 재발 방지 치료를 시작해야 할지 결정하기는 더욱 어렵다. 다만 재발이 있는 환자에서는 장기적인 재발 방지 치료를 시작할 것이 권장된다. 그 외에 치료 여부를 결정하는 데에는 첫 증상 치료시 치료 반응, 첫 증상의 중증도, 장애 축적 위험성, 장기적 면역 치료의 위험성 등을 고려하여야 한다.24 예를 들어, 첫 증상 후 장애 정도가 심하여 적극적으로 재발을 예방해야 하는 경우나 스테로이드 치료에 매우 의존적인 증상 경과를 보이는 경우는 첫 발병이더라도 장기적 면역억제 치료를 고려해볼 수 있겠다. MOG 항체의 음전 여부 또한 재발과 관련이 있기 때문에 주기적으로 MOG항체검사를 하여 면역 치료 여부를 결정하는 데에 참고할 수 있겠다.
장기 재발 방지 치료에 대해서 현재 무작위 대조군 임상 연구가 진행되고 있으나 아직 완료된 연구가 없기 때문에, 후향적 연구, 관찰 연구, 실제임상자료(real world data)를 바탕으로 약물을 살펴보고자 한다. 아자티오프린(azathioprine, 2-3 mg/kg/day), 미코페놀레이트모페틸(mycophenolate mofetil, 2,000 mg/day)과 같은 경구용 면역억제제, 리툭시맙 같은 B세포 억제제, 주기적 면역글로불린 정주(IVIG) 등이 연간 재발률(annualized relapse rate)을 낮추는 것으로 알려졌다. 다만 MS에서 사용하는 질병조절약제(disease modifying drug)는 MOGAD 재발 방지에 효과가 없었다. 아자티오프린, 미코페놀레이트모페틸과 같은 경구용 면역억제제를 사용한 환자에서는 각각 약 39%, 47% 환자에서 재발을 막을 수 있었다.18,23,24,48-51
리툭시맙을 사용한 환자에서는 약 50%에서 재발을 막을 수 있었는데, 이는 AQP4+NMOSD에서의 치료 성적과 비교해서는 효과가 조금 적은 것을 알 수 있다.52 리툭시맙(rituximab)은 CD20을 타깃으로 하는 B세포 억제제로, 시작요법은 1) 375 mg/m2 용량으로 1주마다 4주 동안 투약하거나, 2) 1 g을 2주마다 2회 투약하는 방법이 가능하고, 이후 유지 치료는 375 mg/m2 또는 1 g을 CD19를 모니터하여 1% 초과 시에 또는 6개월마다 투여한다.
주기적 IVIG는 약 69%의 환자에서 재발을 막았으며, 일부 환자들에서는 EDSS 점수의 호전 또한 보고되었다. 일반적으로 4주 간격으로 0.4-2 g/kg의 용량으로 주기적 투여할 수 있다. 몇몇 후향적 연구에서 재발 방지 효과가 다른 면역억제제보다 좋다는 의견이 있으나, 최대 50%의 환자에서 재발이 보고되었고, 특히 낮은 용량일 때 또는 투여 간격이 연장될 때 재발의 위험성이 높은 것으로 보고되었다.
그 외에도 토실리주맙(tocilizumab)은 인터루킨-6 수용체 차단제로 소규모 연구에서 약73% 환자에서 재발을 막았으며, 다른 면역억제 치료에 불응하는 환자에서 8 mg/kg으로 월 1회 투여(최대 800 mg/월)를 시도해볼 수 있겠다.53 하지만 아직 장기 재발 방지 치료를 해야 할 환자의 선별, 치료 기간, 치료 중단에 대한 더 많은 연구가 필요하다.
MOGAD의 임상 경과 및 예후
임상적 재발은 첫 증상 발생 후 6개월 이내가 이후보다 더 잦다. 특히 경구 스테로이드 치료를 줄이거나 중단한 이후 2개월 이내에 자주 재발한다.23,46 성인과 소아에서 재발의 빈도에 차이가 있는데, 소아에서는 재발이 적고 단발형 경과를 보이는 경우가 많다.19,54 여러 장기 추적 코호트에서 5년 이내 재발률은 소아에서 약 17-30%,47,54,55 성인에서는 약 40-62% 정도로 알려져 있다.42,46,56 드물지만 MOGAD 환자에서 특히 첫 증상 발생 후 1년 이내에 무증상 뇌 병변이 추가로 생길 수 있다. 다만 이러한 무증상 뇌 병변이 생기는 경우 향후 재발할 위험성이 높음을 시사할 수 있다.57 MS와는 달리 임상적 재발 없이 질병이 진행하는 것은 매우 드물지만 추가적인 연구가 필요하다.
처음 증상 발생 당시 MOG항체의 역가는 회복이나 재발을 예측할 수 없다. 반복 채혈로 MOG항체를 검사할 경우 지속적인 항체 양성, 특히 높은 역가의 MOG항체를 가진 경우 항체가 음전되는 경우보다 높은 재발률과 연관되어 있었다.
소아에서는 대체로 단발성 경과를 보이는 경우가 많고, EDSS 3점에 도달하는 운동장애가 발생하는 비율 또한 10% 미만(성인에서는 20-40%)으로 예후가 좋은 편이나, 일부 환자에서 백질형성장애 유사 병변으로 발현하는 경우 예후가 좋지 않으니 주의해야 한다. MS와 달리 진행성 경과는 매우 드물고, MS나 AQP4+NMOSD 만큼 재발성 경과를 보이는 경우가 많지는 않지만 일부 환자에서는 빈번한 재발과 반복적인 장애의 축적으로 불량한 예후를 보여주는 경우가 있어 적극적인 재발 방지 치료가 필요하다.
결론
MOG항체의 정확한 검출 방법이 제시되면서부터 MOGAD라는 새로운 진단 범주가 생기고, 이 분야에 활발한 연구가 이루어지고 있다. 병태생리를 밝히고, 발병 기전을 이해하는 것은 병의 경과를 파악하는 바이오마커 발굴 및 치료제 개발에도 중요하다. MOGAD는 임상 양상이 매우 다양하고, 전형적인 MOGAD가 아닌 환자나 건강한 사람에서도 MOG항체가 양성일 수 있기 때문에, MOGAD 표현형에 적합한 환자에서 정확한 방법으로 항체검사를 시행하고 그 결과를 해석하는 것이 위양성 및 오진을 줄이고 검사의 양성예측도를 높이는 데에 중요하다. 아직 치료 효과를 정확하게 판단할 수 있는 연구가 없어, 현재 진행되고 있는 무작위 대조군 임상시험의 결과 또한 기대가 된다. 다만 MS나 AQP4+NMOSD와 같이 모든 환자가 장기 재발 방지 치료가 필요한 것은 아니기 때문에 치료가 필요한 환자를 조기에 선별할 수 있는 방법이 필요하다.