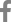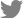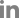ņä£ļĪĀ
Myelin oligodendrocyte glycoprotein (MOG)ņØĆ ņżæņČöņŗĀĻ▓ĮĻ│äņØś ņłśņ┤łņŚÉ ļ░£ĒśäĒĢśļŖö ļŗ╣ļŗ©ļ░▒ņØ┤ļŗż. ņśłņĀäļČĆĒä░ ĒĢŁMOGĒĢŁņ▓┤(anti-MOG antibody, MOGĒĢŁņ▓┤)ņØś ņĪ┤ņ×¼ļŖö ņĢīļĀżņĀĖ ņÖöņ£╝ļéś, Ēśłņ▓ŁņŚÉņä£ ņāØņäĖĒżĻĖ░ļ░śĻ▓Ćņé¼(live cell-based assay)ļź╝ ĒåĄĒĢ£ MOGĒĢŁņ▓┤ņØś Ļ▓ĆņČ£ļ▓ĢņØ┤ ņĢīļĀżņ¦Ćļ®┤ņä£ļČĆĒä░ MOGĒĢŁņ▓┤ņØś ņ×äņāüņĀü ņżæņÜöņä▒ņØ┤ ļīĆļæÉļÉśņŚłĻ│Ā, ņØ┤ļĪ£ ņØĖĒĢ┤ ņżæņČöņŗĀĻ▓ĮĻ│ä ņŚ╝ņ”Ø Ēāłņłśņ┤łņ¦łĒÖś ņżæņŚÉņä£ MOG ĒĢŁņ▓┤ņ¦łĒÖś(MOG antibody-associated disease, MOGAD)ņØ┤ļØ╝ļŖö ļÅģļ”ĮļÉ£ ņ¦łĒÖśņ£╝ļĪ£ņØś ļČäļźśĻ░Ć Ļ░ĆļŖźĒĢśĻ▓ī ļÉśņŚłļŗż.1,2
MOGADļŖö ļŗżļ░£ņä▒Ļ▓ĮĒÖöņ”Ø(multiple sclerosis, MS)ņØ┤ļéś ĒĢŁņĢäņ┐ĀņĢäĒżļ”░4ĒĢŁņ▓┤ ņ¢æņä▒ ņŗ£ņŗĀĻ▓Įņ▓ÖņłśņŚ╝ļ▓öņŻ╝ņ¦łĒÖś(anti-aquaporin 4 antibody positive neuromyelitis optica spectrum disorder, AQP4+NMOSD)Ļ│╝ļŖö ņżæņČöņŗĀĻ▓ĮĻ│ä ņŚ╝ņ”Ø Ēāłņłśņ┤łņ¦łĒÖśņØ┤ļØ╝ļŖö Ļ│ĄĒåĄņĀÉņØ┤ ņ׳ņ¦Ćļ¦ī, ņ×äņāü Ļ▓ĮĻ│╝, ļ│æļ”¼ĒĢÖņĀü ĒŖ╣ņ¦Ģ, ļ│æĒā£ņāØļ”¼, ņśüņāüņØśĒĢÖņĀü ĒŖ╣ņ¦Ģ, ņ╣śļŻī ļ░śņØæ, ņśłĒøä ļō▒ ļ¦ÄņØĆ ļČĆļČäņŚÉņä£ ņ░©ņØ┤ņĀÉņØ┤ ņ׳ļŗż.2-6 ļ¬©ļōĀ ņŚ░ļĀ╣ļīĆņŚÉņä£ ļ░£ņāØĒĢĀ ņłś ņ׳ņ£╝ļ®░, ņĀä ņäĖĻ│äņĀüņ£╝ļĪ£ ņ£Āļ│æļźĀņØĆ 10ļ¦ī ļ¬ģļŗ╣ 2.0ļ¬ģ, ļ░£ļ│æļźĀņØĆ ņŚ░Ļ░ä 10ļ¦ī ļ¬ģļŗ╣ 0.2-0.3ļ¬ģņ£╝ļĪ£ ņČöņĀĢļÉ£ļŗż.7,8 ņä▒ņØĖ ņżæņČöņŗĀĻ▓ĮĻ│ä ņŚ╝ņ”Ø Ēāłņłśņ┤łņ¦łĒÖś ņżæņŚÉļŖö ņĢĮ 1-6%ļź╝ ņ░©ņ¦ĆĒĢśĻ│Ā, ņåīņĢäņŚÉņä£ļŖö ĻĖēņä▒ Ēāłņłśņ┤łņ¦łĒÖśņØś ņĢĮ 40% ņĀĢļÅäĻ╣īņ¦Ć ļ│┤Ļ│ĀļÉ£ļŗż. ņ£Āļ│æļźĀņØĆ ņĀÉņ░© ņ”ØĻ░ĆĒĢśĻ│Ā ņ׳Ļ│Ā, ņĄ£ĻĘ╝ MOGADņØś ĻĄŁņĀ£ ņ¦äļŗ© ĻĖ░ņżĆ3ņØ┤ ņĀ£ņŗ£ļÉ©ņŚÉ ļö░ļØ╝ Ē¢źĒøä ļŹö ņ£ĀļÅÖņĀüņØ╝ Ļ▓āņ£╝ļĪ£ ņśłņāüļÉ£ļŗż.
ļ│Ė ņóģņäżņŚÉņä£ļŖö MOGADņØś ļ│æĒā£ņāØļ”¼, ņ×äņāü ņ¢æņāü, ņ×äņāü Ļ▓ĮĻ│╝, Ļ▓Ćņé¼ļ▓ĢĻ│╝ ņ¦äļŗ© ĻĖ░ņżĆ ļ░Å ņ╣śļŻīņÖĆ ņśłĒøäņŚÉ ļīĆĒĢ£ ņĀäļ░śņĀüņØĖ ļé┤ņÜ®ņØä ņé┤ĒÄ┤ļ│┤Ļ│Āņ×É ĒĢ£ļŗż.
ļ│Ėļ¼Ė
MOGADņØś ļ│æĒā£ņāØļ”¼
MOGĒĢŁņ▓┤ņØś ņāØņä▒ņØä ņ£Āļ░£ĒĢśļŖö ņØĖņ×ÉļŖö ļ¬ģĒÖĢĒ׳ ņĢīļĀżņ¦Ćņ¦Ć ņĢŖņĢśņ¦Ćļ¦ī ļ¦Éņ┤łļ®┤ņŚŁĻ│ä(peripheral immune system)ņŚÉņä£ ņ×ÉĻ░Ćļ®┤ņŚŁ ĻĖ░ņĀäņŚÉ ņØśĒĢ┤ ņāØĻĖ┤ļŗżĻ│Ā ņŚ¼Ļ▓©ņ¦ĆĻ│Ā ņ׳ļŗż.9 MOGĒĢŁņ▓┤ ņāØņä▒ņŚÉļŖö BņäĖĒżļéś ĒśĢņ¦łņäĖĒż(plasma cell) ņÖĖņŚÉļÅä, BņäĖĒżĻ░Ć ĒśĢņ¦łņäĖĒżļĪ£ ļČäĒÖöĒĢśļÅäļĪØ ļÅäņÖĆņŻ╝ļŖö ĒĢŁņ▓┤ĒŖ╣ņØ┤ņĀü ņŚ¼Ēżļ│┤ņĪ░TņäĖĒż(antigen-specific T follicular helper cell)Ļ░Ć ņŚ░Ļ┤ĆļÉśņ¢┤ ņ׳ļŗżĻ│Ā ņČöņĖĪļÉ£ļŗż. MOGĒĢŁņ▓┤Ļ░Ć ļ│æņØä ņØ╝ņ£╝ĒéżĻĖ░ ņ£äĒĢ┤ņä£ļŖö ņżæņČöņŗĀĻ▓ĮĻ│ä ļé┤ļĪ£ ņ£Āņ×ģļÉśņ¢┤ņĢ╝ ĒĢ£ļŗż. ĒśłņĢĪļćīņןļ▓Į(blood-brain barrier, BBB)ņØ┤ ņåÉņāüļÉśņ¢┤ ĒÖ£ņä▒ĒÖöļÉ£ BņäĖĒż, ĒśĢņ¦łņäĖĒż ļ░Å MOGĒĢŁņ▓┤Ļ░Ć BBBļź╝ ĒåĄĻ│╝ĒĢśļ®┤, ņżæņČöņŗĀĻ▓ĮĻ│ä ļé┤ņŚÉņä£ļŖö MOGĒĢŁņ▓┤Ļ░Ć MOGļź╝ ļ░£ĒśäĒĢśļŖö ņłśņ┤łņŚÉ Ļ▓░ĒĢ®ĒĢśņŚ¼ ņłśņ┤łņåÉņāü ļ░Å Ēāłņłśņ┤łļź╝ ņ¦äĒ¢ēņŗ£Ēé©ļŗż. ņØ┤ļĢī, ļīĆņŗØņäĖĒż(macrophage)ņÖĆ TņäĖĒż(ņŻ╝ļĪ£ CD4+ TņäĖĒż), Ļ│╝ļ”ĮĻĄ¼(granulocyte) ļō▒ņØ┤ Ļ░ÖņØ┤ ņżæņČöņŗĀĻ▓ĮĻ│ä ļé┤ļĪ£ ņ£Āņ×ģļÉśļ®░, ņé¼ņŗż ļ│æļ│ĆņŚÉļŖö ņØ┤ļōż ņäĖĒżĻ░Ć BņäĖĒżļ│┤ļŗż ļŹö ļ¦ÄņØĆ ļ╣äņ£©ņØä ņ░©ņ¦ĆĒĢśĻ│Ā ņ׳ņ¢┤ ļ│┤ņ▓┤ ĒÖ£ņä▒ņØä ņØ╝ņ£╝ĒéżĻĖ░ļÅä ĒĢ£ļŗż.10,11 ņØ┤ļĪ£ ņØĖĒĢ┤ MOGADņØś ļ│æļ”¼ĒĢÖņĀü ņåīĻ▓¼ņØĆ ņĀĢļ¦źņŻ╝ņ£ä ĒĢ®ļźśņä▒ Ēāłņłśņ┤ł ļ│æļ│Ć, Ēö╝ņ¦łļé┤ Ēāłņłśņ┤ł ļ│æļ│Ć, ļ│æļ│Ć ļé┤ MOG ļ░£Ēśä ņłśņ┤ł ņåīņŗż, CD4+ TņäĖĒż ļ░Å Ļ│╝ļ”ĮĻĄ¼ ņÜ░ņäĖ ņŚ╝ņ”Ø, ĒÖ£ļÅÖņä▒ ļ░▒ņ¦ł ļ│æļ│Ć ļé┤ ļ│┤ņ▓┤ņ╣©ņ░®, ļČĆļČäņĀü ņČĢņéŁ ļ│┤ņĪ┤ ļō▒ņ£╝ļĪ£ ĒŖ╣ņ¦Ģņ¦ĆņØä ņłś ņ׳ļŗż. MSņÖĆļŖö ļŗ¼ļ”¼ CD8+ TņäĖĒżļ│┤ļŗż CD4+ TņäĖĒżĻ░Ć ņÜ░ņäĖĒĢśĻ│Ā, ņä£ņä£Ē׳ ĒÖĢņןļÉśļŖö ĒöīļØ╝Ēü¼(slowly expending plaque)Ļ░Ć ņŚåņ£╝ļ®░, AQP4+NMOSDņÖĆļŖö ļŗ¼ļ”¼ AQP4Ļ░Ć ņל ļ│┤ņĪ┤ļÉśņ¢┤ņ׳Ļ│Ā, AQP4 ņŻ╝ļ│ĆņØś ļ│┤ņ▓┤ ņ╣©ņ░®ņØ┤ Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖļŖöļŗż.5,6 ļśÉĒĢ£ ņĀäņŚ╝ņ”Øņä▒ ņé¼ņØ┤ĒåĀņ╣┤ņØĖ(proinflammatory cytokine) ļ░Å BņäĖĒż ņé¼ņØ┤ĒåĀņ╣┤ņØĖ/ņ╝Ćļ¬©ņ╣┤ņØĖņØ┤ ņ”ØĻ░ĆļÉ©ņØ┤ ļćīņ▓ÖņłśņĢĪ ļśÉļŖö ĒśłņĢĪņŚÉņä£ ĒÖĢņØĖļÉśņŚłļŗż.12,13
MOGADņØś ņ×äņāü ņ¢æņāü
MOGADļŖö MSļéś AQP4+NMOSDņÖĆ Ļ░ÖņØ┤ ņŚ¼ņä▒ņŚÉņä£ ņÜ░ņäĖĒĢśņ¦ĆļŖö ņĢŖĻ│Ā, ņĀä ņŚ░ļĀ╣ļīĆņŚÉņä£ ļ░£ņāØĒĢĀ ņłś ņ׳ņ£╝ļéś ņä▒ņØĖĻ│╝ ņåīņĢäņŚÉņä£ ļ░£ĒśäĒĢśļŖö ņ×äņāü ņ¢æņāü Ēæ£ĒśäĒśĢņŚÉ ņ░©ņØ┤Ļ░Ć ņ׳ļŗż(Table 1).14,15 ņä▒ņØĖņŚÉņä£ļŖö ņŗ£ņŗĀĻ▓ĮņŚ╝(optic neuritis)ņØ┤, ņåīņĢäņŚÉņä£ļŖö ĻĖēņä▒Ēīīņóģļćīņ▓ÖņłśņŚ╝(acute disseminated encephalomyelitis, ADEM)ņØ┤ Ļ░Ćņן ĒØöĒĢ£ ņ×äņāüņØ┤ļŗż. ĒŖ╣Ē׳ ņåīņĢäņŚÉņä£ļŖö ņĀäņ▓┤ ADEM ĒÖśņ×É ņżæ ļ¦ÄĻ▓īļŖö 64%Ļ╣īņ¦Ć, ĻĘĖļ”¼Ļ│Ā ņ×¼ļ░£ Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖö ADEM ĒÖśņ×ÉļŖö ļīĆļČĆļČä MOGADļĪ£ ņ¦äļŗ©ļÉ£ļŗż.16,17 ĻĘĖ ņÖĖņŚÉ ņ▓ÖņłśņŚ╝ ļŗ©ļÅģ ļśÉļŖö ņŗ£ņŗĀĻ▓ĮņŚ╝Ļ│╝ ņ▓ÖņłśņŚ╝ņØ┤ ļÅÖļ░śļÉśļŖö Ļ▓ĮņÜ░ ļśÉĒĢ£ ĒØöĒĢśĻ▓ī Ļ┤Ćņ░░ļÉśļ®░, ĒŖ╣Ē׳ AQP4 ĒĢŁņ▓┤ ņØīņä▒ņØĖ NMOSD ļśÉļŖö ņóģļŗ©Ļ┤æļ▓öņ£äĒÜĪļŗ©ņ▓ÖņłśņŚ╝(longitudinally extensive transverse myelitis, LETM)ņØś ņāüļŗ╣ņłśņŚÉņä£ MOGADļĪ£ ņ¦äļŗ©ļÉśĻĖ░ļÅä ĒĢ£ļŗż.18,19 ĻĘĖ ņÖĖņŚÉ ņØ╝ļČĆ ĒÖśņ×ÉļōżņØĆ ļīĆļćīĒö╝ņ¦łļćīņŚ╝, ļćīĻ░ä ļ░Å ņåīļćī ļ│æļ│Ć, ņóģĻ┤┤ĒśĢņä▒ Ēāłņłśņ┤ł ļ│æļ│Ć, ļćīņŗĀĻ▓Įļ│æņ”Ø, ņ¦äĒ¢ēņä▒ ļ░▒ņ¦ł ņåÉņāü ļō▒ņ£╝ļĪ£ ļ░£ĒśäĒĢ£ļŗż. ņ╣©ļ▓ö ļČĆņ£äņŚÉ ļö░ļźĖ ņ×äņāüņĀü ĒŖ╣ņ¦ĢņØĆ ļŗżņØīĻ│╝ Ļ░Öļŗż.
ņŗ£ņŗĀĻ▓Į ļ│æļ│Ć
MOGAD ĒÖśņ×ÉņØś ņĢĮ 80%Ļ╣īņ¦Ć ņŗ£ņŗĀĻ▓ĮņØ┤ ņ╣©ļ▓öļÉĀ ņłś ņ׳ņ£╝ļ®░, ĒŖ╣Ē׳ ņ¢æņĖĪņä▒(31-58%), ņ×¼ļ░£ņä▒ņ£╝ļĪ£ ļ░£ņāØĒĢĀ ņłś ņ׳ļŗż.20,21 ļīĆĒæ£ņĀüņØĖ ņ”Øņāü/ņ”ØĒøäļĪ£ļŖö ņżæņŗ¼ņŗ£ļĀźņĀĆĒĢś, ņĢłĻĄ¼ļÆżĒåĄņ”Ø, ņāēĻ░üņØ┤ņāü, ĻĄ¼ņŗ¼ļÅÖĻ│ĄĻ▓░ņåÉ(afferent pupillary defect) ļō▒ņØ┤ ņ׳Ļ│Ā, Ļ▓Ćņ¦äņŚÉņä£ļŖö ņŗ£ņŗĀĻ▓Įņ£ĀļæÉ ļČĆņóģņØ┤ ĒØöĒ׳ Ļ┤Ćņ░░ļÉ£ļŗż. ņŗĀĻ▓ĮņāØļ”¼Ļ▓Ćņé¼ņŚÉņä£ļŖö ņŗ£Ļ░üņ£Āļ░£ņĀäņ£ä(visual evoked potential), Ļ┤æĻ░äņäŁļŗ©ņĖĄņ┤¼ņśü(optical coherence tomography, OCT) ļō▒ņØś Ļ▓Ćņé¼ņŚÉ ņØ┤ņāüņØä ļ│┤ņØ╝ ņłś ņ׳ņ£╝ļéś ņ¦äļŗ©ņŚÉ ĒŖ╣ņØ┤ņĀüņØ┤ņ¦ĆļŖö ņĢŖļŗż. ņŗ£ņŗĀĻ▓ĮņŚ╝ ļŗ©ļÅģņ£╝ļĪ£ļÅä ļéśĒāĆļéśņ¦Ćļ¦ī, ADEMņØ┤ļéś ņ▓ÖņłśņŚ╝Ļ│╝ ļÅÖļ░śļÉśņ¢┤ ļéśĒāĆļéśĻĖ░ļÅä ĒĢ£ļŗż.
ņŗ£ļĀźņĀĆĒĢśļŖö ĒÖśņ×Éļ¦łļŗż ņ░©ņØ┤Ļ░Ć ņ׳ņ¦Ćļ¦ī ņóģņóģ ļ¦żņÜ░ ņŗ¼ĒĢśĻ▓ī ļéśĒāĆļéśĻĖ░ļÅä ĒĢ£ļŗż. ļŗżļ¦ī ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻīņŚÉ ļ░śņØæņØ┤ ņóŗņĢä ļ╣Āļź┤Ļ▓ī ĒÜīļ│ĄļÉĀ ņłś ņ׳ņ£╝ļéś, ņŖżĒģīļĪ£ņØ┤ļō£ļź╝ ņżäņØ┤Ļ▒░ļéś ņżæļŗ©ĒĢ£ ļÆż ļ░öļĪ£ ņ×¼ļ░£ĒĢĀ ņłśļÅä ņ׳ļŗż.22,23 ņĢĮ 30-50%ņØś MOG ĒĢŁņ▓┤ ņ¢æņä▒ ņŗ£ņŗĀĻ▓ĮņŚ╝ņØĆ ņ×¼ļ░£ĒĢśļŖö Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖöļŹ░, ņ▓śņØīņŚÉļŖö ADEMņØ┤ļéś NMOSDņŚÉ ļÅÖļ░śĒĢśņŚ¼ ļ░£ļ│æĒĢśņśĆļŗżĻ░Ć ņŗ£ņŗĀĻ▓ĮņŚ╝ņ£╝ļĪ£ ņ¦ĆņåŹņĀüņ£╝ļĪ£ ņ×¼ļ░£ĒĢśĻĖ░ļÅä ĒĢ£ļŗż.24
ņ¦äļŗ©ņØä ņ£äĒĢ£ ņĢłĻĄ¼ MRI ņ┤¼ņśü ņŗ£ņŚÉļŖö T2Ļ░ĢņĪ░ņśüņāü ļ░Å T1ņĪ░ņśüņ”ØĻ░ĢņśüņāüņŚÉņä£ Ļ│ĀņŗĀĒśĖĻ░ĢļÅäļź╝ ļ│┤ņØ┤ļ®░, ĒŖ╣Ē׳ ņ¦Ćļ░®ņ¢ĄņĀ£(fat-suppression)ĻĖ░ļ▓ĢņØä ņØ┤ņÜ®ĒĢ£ ņĪ░ņśüņ”ØĻ░ĢņśüņāüņØä ņ┤¼ņśüĒĢśļŖö Ļ▓āņØ┤ ņŗ£ņŗĀĻ▓Į ņŚ╝ņ”Ø ĒīÉļŗ©ņŚÉ ņ£Āļ”¼ĒĢśļŗż(Fig. 1A). MS ļśÉļŖö AQP4+NMOSDņÖĆņØś ņ░©ņØ┤ņĀÉņ£╝ļĪ£ļŖö ņŗ£ņŗĀĻ▓Į ņżæ ņĢłņÖĆ(orbital) ļČĆņ£äļź╝ ņ×ÉņŻ╝ ņ╣©ļ▓öĒĢśņŚ¼ ņŗ£ņŗĀĻ▓ĮņØś ļ©Ėļ”¼ļČĆļČä ļČĆņóģņØ┤ ļéśĒāĆļéĀ ņłś ņ׳Ļ│Ā, ņØ┤ļĪ£ ņØĖĒĢ£ ņĢłĻĄ¼ļÆżĒåĄņ”ØņØ┤ ļŹö ĒśĖļ░£ĒĢśĻĖ░ļÅä ĒĢ£ļŗż. ņŗ£ņŗĀĻ▓Į ļ│æļ│ĆņØĆ ĻĖĖĻ▓ī ņ╣©ļ▓öĒĢśļŖö Ļ▓āņØ┤ ĒŖ╣ņ¦ĢņĀüņØ┤ļ®░, ņŗ£ņŗĀĻ▓Įņ¦æ(optic nerve sheath)ņØä ņ╣©ļ▓öĒĢśņŚ¼ ņĪ░ņśüņ”ØĻ░ĢņØä ļéśĒāĆļé╝ ņłś ņ׳ļŗż.18,25 ļśÉĒĢ£, ņŗ£ņŗĀĻ▓Įņ£ĀļæÉļČĆņóģ ļ░Å ņČ£ĒśłņØ┤ ļŹö ĒØöĒĢśĻ▓ī Ļ┤Ćņ░░ļÉĀ ņłś ņ׳ņ¢┤ ņØ┤ļź╝ ĒÖĢņØĖĒĢĀ ņłś ņ׳ļŖö ņĢłņĀĆĻ▓Ćņé¼Ļ░Ć ņ£ĀņÜ®ĒĢśļŗż. ņŗ£ņŗĀĻ▓ĮņŚ╝ ĻĖēņä▒ĻĖ░ ļĢī OCTļĪ£ ņŗ£ņŗĀĻ▓Įņ£ĀļæÉņŻ╝ņ£äļ¦Øļ¦ēņŗĀĻ▓Įņä¼ņ£ĀņĖĄ(peripapillary retinal nerve fiber layer)ņØ┤ ļæÉĻ║╝ņøīņ¦ä Ļ▓āņØä ĒÖĢņØĖĒĢĀ ņłś ņ׳Ļ│Ā, ņØ┤ļŖö ĻĖēņä▒ĻĖ░Ļ░Ć ņ¦Ćļéśļ®┤ ņĀÉņ░© ņ¢ćņĢäņ¦ĆļŖöļŹ░, ĒŖ╣Ē׳ ņĖĪļæÉņé¼ļČäļ®┤(temporal quadrant)ņØ┤ ļŹö ņŗ¼ĒĢśĻ▓ī ņ¢ćņĢäņ¦äļŗż. ņŗ£ņŗĀĻ▓ĮņŚ╝ņŚÉņä£ļŖö ĒÖ®ļ░śļČĆ ņŗĀĻ▓ĮņĀłņäĖĒż-ļé┤ļ¦ØņāüņĖĄ(ganglion cell-inner plexiform layer)ņØś ļæÉĻ╗ś ļśÉĒĢ£ ņ¢ćņĢäņ¦ł ņłś ņ׳ļŖöļŹ░, MOGADņÖĆ Ļ┤ĆļĀ©ļÉ£ ņŗ£ņŗĀĻ▓ĮņŚ╝ņØĆ AQP4+NMOSDļéś MSņÖĆ ļ╣äĻĄÉĒĢ┤ņä£ ĻĘĖ ņĀĢļÅäĻ░Ć Ļ▓Įļ»ĖĒĢśņŚ¼ ļ¦Øļ¦ēņØś ņåÉņāüņØ┤ ļŹ£ĒĢśļŗżĻ│Ā ņ£ĀņČöĒĢĀ ņłś ņ׳ļŗż.26
ļćī ļ░Å ļćīĻ░ä ļ│æļ│Ć
MOGAD ĒÖśņ×ÉņŚÉņä£ ļćī/ļćīĻ░äņØä ņ╣©ļ▓öĒĢśļŖö ĒÖśņ×ÉļŖö ADEM, ļīĆļćīĒö╝ņ¦łļćīņŚ╝, ļćīĻ░ä ļ░Å ņåīļćīņ”ØĒøäĻĄ░ ļō▒ņØś ņ¢æņāüņ£╝ļĪ£ ļ░£ĒśäĒĢ£ļŗż. ļ¼┤ņ”Øņāü ļćīļ│æļ│ĆļÅä ņ׳ņ¦Ćļ¦ī, ņŗ£ņŗĀĻ▓ĮņŚ╝ņØ┤ļéś ņ▓ÖņłśņŚ╝ņ£╝ļĪ£ ņ▓śņØī ļ░£ĒśäĒĢ£ ĒÖśņ×ÉņŚÉņä£ ļćī ļ│æļ│ĆņØ┤ ņŚåļŖö Ļ▓ĮņÜ░Ļ░Ć ņĢĮ 47-68%ļĪ£ ļŹö ļ¦ÄņĢä MSņÖĆ ĻĄ¼ļ│äļÉĀ ņłś ņ׳ļŗż.4 ļćīļ│æļ│ĆņØĆ ņ¢æņĖĪņä▒, Ļ▓ĮĻ│äĻ░Ć ļČłļ¬ģĒÖĢĒĢ£(ill-defined), Ēü░ ļ│æļ│ĆņØ┤ ļ¦ÄĻ│Ā(Fig. 1B), Ļ░äĒś╣ ņŗ¼ļČĆĒÜīņāēņ¦ł(deep gray matter)ņØä ņ╣©ļ▓öĒĢśĻĖ░ļÅä ĒĢ£ļŗż. ņĀäĒśĢņĀüņØĖ MS ļ│æļ│ĆņØ┤ļéś ņ¦ĆņåŹņĀüņØĖ T1 ņĀĆĻ░ĢļÅäņŗĀĒśĖ ļ│æļ│ĆņØĆ MOGADņŚÉņä£ļŖö ļō£ļ¼╝ļŗż. ļćīĻ░ä ļ│æļ│ĆņØś Ļ▓ĮņÜ░ ĻĄÉļćīņ╣©ļ▓öņØ┤ ĒØöĒĢśļ®░, ĒŖ╣Ē׳ ņżæĻ░äņåīļćīļŗżļ”¼ļź╝ ņ╣©ļ▓öĒĢśļŖö Ēü░ ļ│æļ│ĆņØś Ļ▓ĮņÜ░ņŚÉļŖö MSļéś AQP4+NMOSDļ│┤ļŗż MOGADļź╝ ņŗ£ņé¼ĒĢśĻ│Ā,27 ļŗżļ¦ī ļ¦©ņĢäļלĻĄ¼ņŚŁņ”ØĒøäĻĄ░(area postrema syndrome)ņ£╝ļĪ£ ļéśĒāĆļéśļŖö Ļ▓ĮņÜ░ļŖö ļ¦żņÜ░ ļō£ļ¼╝ļŗż.28 ņåīņĢäņŚÉņä£ļŖö MOGAD ĒÖśņ×ÉņØś ņĢĮ 50%ļź╝ ņ░©ņ¦ĆĒĢĀ ņĀĢļÅäļĪ£ ADEMņØ┤ Ļ░Ćņן ĒØöĒĢśņ¦Ćļ¦ī ņä▒ņØĖņŚÉņä£ļŖö ņĢĮ 5.6%ļĪ£ ļ¦Äņ¦Ć ņĢŖļŗż.19 ĒŖ╣Ē׳ ņåīņĢä MOGADļŖö ADEM ņĀäņŚÉ Ļ░ÉņŚ╝ņä▒ ņ¦łĒÖśņØ┤ ņäĀĒ¢ēĒĢśļŖö Ļ▓ĮņÜ░Ļ░Ć 40-75%ļĪ£ ļ¦ÄĻ│Ā, ņĢĮ 70% ĒÖśņĢäļŖö ņśüņāüņØśĒĢÖņĀüņ£╝ļĪ£ Ļ┤ĆĒĢ┤Ļ░Ć ļÉ£ļŗż.29 ĒĢśņ¦Ćļ¦ī ļ░▒ņ¦łĒśĢņä▒ņןņĢĀ ņ£Āņé¼ ļ│æļ│Ć(leukodystrophy like lesion) ņ¢æņāüņ£╝ļĪ£ ļéśĒāĆļéśļŖö Ļ▓ĮņÜ░ļŖö ņśłĒøäĻ░Ć ļČłļ¤ēĒĢśĻ│Ā, MOGĒĢŁņ▓┤Ļ░Ć ņ¢æņä▒ņØĖ ADEMņØś Ļ▓ĮņÜ░ ņØīņä▒ņØĖ Ļ▓ĮņÜ░ļ│┤ļŗż ņ×¼ļ░£ņØä ņל ĒĢ£ļŗż. ļīĆļćī ļćīņŚ╝ņØĆ ļ░£ņŚ┤, ļæÉĒåĄ, ņØśņŗØņĀĆĒĢś, ļ░£ņ×æ ļō▒ņØś ņ”ØņāüņØä ļ│┤ņØ┤ļ®░, ņØ┤ ņżæ ļ░£ņ×æĻ│╝ ĒĢ©Ļ╗ś ļīĆļćī Ēö╝ņ¦łņŚÉ ņĢĪņ▓┤Ļ░ÉņćĀņŚŁņĀäĒÜīļ│Ąņśüņāü(fluid attenuated inversion recovery, FLAIR) MRIņŚÉņä£ Ļ│ĀņŗĀĒśĖĻ░ĢļÅäļź╝ ļ│┤ņØ┤ļŖö ļ│æļ│ĆņØ┤ ņ׳ļŖö Ļ▓ĮņÜ░ļź╝ FLAMES (FLAIR hyperintense lesions in anti-MOG encephalitis with seizures)ļĪ£ ļ¬ģņ╣ŁĒĢśņśĆļŗż(Fig. 1C).30 ĻĘĖ ņÖĖņŚÉ, NMDAņłśņÜ®ņ▓┤ ĒĢŁņ▓┤ņÖĆ MOGĒĢŁņ▓┤Ļ░Ć ļÅÖņŗ£ņŚÉ ņ¢æņä▒ņØĖ ļćīņŚ╝ļÅä ņ׳ļŗż.31,32
ņ▓Öņłś ļ│æļ│Ć
ņ▓Öņłś ļ│æļ│ĆņØĆ ņ▓ÖņłśņŚ╝ ļŗ©ļÅģ ļśÉļŖö ADEMņØ┤ļéś ņŗ£ņŗĀĻ▓ĮņŚ╝Ļ│╝ ļÅÖļ░śĒĢśņŚ¼ ļéśĒāĆļéĀ ņłś ņ׳ļŗż. Ļ░ÉĻ░üņØ┤ņāü, ĻĘ╝ņ£äņĢĮ, Ļ┤äņĢĮĻĘ╝ņןņĢĀ, ņä▒ĻĖ░ļŖźņןņĢĀ ļō▒ņØ┤ ļ░£ņāØĒĢĀ ņłś ņ׳ņ£╝ļ®░, ņ”Øņāü ņżæņ”ØļÅäļŖö 50% ņØ┤ņāüņØś ĒÖśņ×ÉĻ░Ć ĒÖĢņןņןņĢĀņāüĒā£ņ▓ÖļÅä(Expanded Disability Status Scale, EDSS) 4ņĀÉņØä ņ┤łĻ│╝ĒĢśļŖö ņżæļō▒ļÅä ļśÉļŖö ņŗ¼ĒĢ£ ņ”Øņāüņ£╝ļĪ£ ļéśĒāĆļéśņ¦Ćļ¦ī ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻīņŚÉ ļīĆļČĆļČä ņÜ┤ļÅÖĻĖ░ļŖź ĒÜīļ│ĄņŚÉļŖö ļ░śņØæņØ┤ ņóŗļŗż. ļ░░ļć© ļ░Å ļ░░ļ│ĆņןņĢĀļéś ņä▒ĻĖ░ļŖźņןņĢĀļŖö ņ¦ĆņåŹņĀüņ£╝ļĪ£ ļéśĒāĆļéĀ ņłś ņ׳Ļ│Ā, ĒåĄņ”Øņä▒ ĻĖ┤ņןņŚ░ņČĢ(painful tonic spasm)ņØĆ AQP4+NMOSDļ¦īĒü╝ ĒØöĒĢśņ¦ĆļŖö ņĢŖļŗż.33,34
MRI ņ┤¼ņśü ņŗ£ T2Ļ░ĢņĪ░ņśüņāüņŚÉņä£ Ļ│ĀņŗĀĒśĖĻ░ĢļÅäļź╝ ļ│┤ņØ┤ļŖö ļ│æļ│ĆņØ┤ ļ│┤ņØ┤ļéś, ņĢĮ 10% ņĀĢļÅäļŖö ņ▓½ ņ”Øņāü ļ░£ņāØ ņŗ£ĻĖ░ņŚÉ MRIĻ░Ć ņĀĢņāüņØ╝ ņłś ņ׳ļŗż.35 ļ│æļ│ĆņØĆ ļŗ©ļÅģ ļśÉļŖö ļŗżņłśņØś LETMņ£╝ļĪ£ ļéśĒāĆļéśļŖö Ļ▓ĮņÜ░Ļ░Ć 60% ņĀĢļÅäļĪ£ ļ¦ÄĻ│Ā, ņĢĮ 30-50%ņŚÉņä£ļŖö ĒÜīņāēņ¦łņØä ņ╣©ļ▓öĒĢśņŚ¼ H-signņØ┤ ļ│┤ņØ┤Ļ▒░ļéś ņ▓ÖņłśņøÉļ┐öņØä ņ╣©ļ▓öĒĢśļŖö Ļ▓ĮņÜ░ļÅä ņĢĮ 26% ņĀĢļÅäļĪ£ MSļéś AQP4+NMOSDņÖĆ ļ╣äĻĄÉĒĢśņŚ¼ ĒŖ╣ņ¦ĢņĀüņØ┤ļŗż(Fig. 1D).33 ļīĆļČĆļČä ņ▓ÖņłśņØś T2Ļ│ĀņŗĀĒśĖĻ░ĢļÅäļŖö ņŗ£Ļ░äņØ┤ ņ¦Ćļéśļ®┤ ņżäņ¢┤ļōżĻ▒░ļéś ņŚåņ¢┤ņ¦ĆĻĖ░ļÅä ĒĢśņ¦Ćļ¦ī, ņŗ¼ĒĢ£ ņ▓Öņłśļ│æņØĖ Ļ▓ĮņÜ░ ņ▓Öņłś ņ£äņČĢņØ┤ ļéśĒāĆļéśļŖö Ļ▓ĮņÜ░ļÅä ņ׳ļŗż.34,36 ņŗ£ņŗĀĻ▓ĮņØ┤ļéś ļćīļ│æļ│Ć ņ”ØņāüņØä ļ│┤ņØ┤ļŖö ĒÖśņ×ÉņŚÉņä£ļÅä ļ¼┤ņ”Øņāü ņ▓Öņłś ļ│æļ│ĆņØ┤ ļ░£Ļ▓¼ļÉśĻĖ░ļÅä ĒĢśļ®░, ņ▓Öņłśļ│æ ņ”Øņāüņ£╝ļĪ£ ļ░£ĒśäĒĢśļŹöļØ╝ļÅä ļćī ļśÉļŖö ņŗ£ņŗĀĻ▓ĮņŚÉ ļ¼┤ņ”Øņāü ļ│æļ│ĆņØ┤ ļ░£Ļ▓¼ļÉśļŖö Ļ▓ĮņÜ░ļÅä ņĢĮ 33-50% ņĀĢļÅä ļÉ£ļŗż.
ļćīņ▓ÖņłśņĢĪĻ▓Ćņé¼
ļćīņŚ╝ņØś Ļ▓ĮņÜ░ ļæÉĻ░£ļé┤ņĢĢ ņāüņŖ╣ņØ┤ ļÅÖļ░śļÉĀ ņłś ņ׳ņ£╝ļ®░, ĻĖēņä▒ĻĖ░ ļćīņ▓ÖņłśņĢĪĻ▓Ćņé¼ņŚÉņä£ 50% ņØ┤ņāüņØś ĒÖśņ×ÉĻ░Ć ļćīņ▓ÖņłśņĢĪņäĖĒżņ”ØĻ░Ćņ”Ø(>5 WBC/mm3)ņØä ļ│┤ņØ┤Ļ│Ā, ĒŖ╣Ē׳ ņØ┤ ņżæ 12% ņĀĢļÅäļŖö ņŗ¼ĒĢ£ ļćīņ▓ÖņłśņĢĪ ņäĖĒżņ”ØĻ░Ćņ”Ø(>100 WBC/mm3)ņØä ļ│┤ņØ┤ĻĖ░ļÅä ĒĢśļŖöļŹ░, ņŗ£ņŗĀĻ▓ĮņŚ╝ļ│┤ļŗż ADEMņØ┤ļéś ņ▓ÖņłśņŚ╝ ĒÖśņ×ÉņŚÉņä£ ļŹö ĒØöĒĢśĻ▓ī Ļ┤Ćņ░░ļÉ£ļŗż. ĻĘĖļ”¼Ļ│Ā ņĢĮ 30% ĒÖśņ×ÉņŚÉņä£ ļćīņ▓ÖņłśņĢĪ ļŗ©ļ░▒ ņ”ØĻ░ĆĻ░Ć ļéśĒāĆļé£ļŗż. ļćīņ▓ÖņłśņĢĪņŚÉņä£ ņś¼ļ”¼Ļ│ĀĒü┤ļĪĀļØĀ(oligoclonal band)ļŖö ļīĆļČĆļČä ņØīņä▒ņØ┤ļéś ņĢĮ 20%Ļ╣īņ¦Ć ņ¢æņä▒ņ£╝ļĪ£ ļ│┤Ļ│ĀļÉĀ ņłś ņ׳ņ£╝ļ®░, ņØ╝ņŗ£ņĀüņ£╝ļĪ£ ļéśĒāĆļéśĻĖ░ļÅä ĒĢ£ļŗż.37,38
MOGADņØś ņ¦äļŗ©
MOGĒĢŁņ▓┤Ļ▓Ćņé¼
MOGĒĢŁņ▓┤Ļ▓Ćņé¼ļŖö Ēśłņ▓ŁņŚÉņä£ ņŗ£Ē¢ēĒĢśļŖö Ļ▓āņØä ņÜ░ņäĀņ£╝ļĪ£ ĒĢśļ®░, Ļ▓ĮņÜ░ņŚÉ ļö░ļØ╝ņä£ļŖö ļćīņ▓ÖņłśņĢĪņŚÉņä£ ņŗ£Ē¢ēĒĢĀ ņłś ņ׳ļŗż.1,3 Ēśłņ▓Ł MOGĒĢŁņ▓┤ Ļ▓Ćņé¼ļŖö ņØĖĻ░ä MOGņØś ņĀäņ▓┤ ĻĖĖņØ┤ņØś ļŗ©ļ░▒ņ¦łņØä ņØ┤ņÜ®ĒĢ£ ņāØņäĖĒżĻĖ░ļ░ś ļČäņäØ(live cell-based assay)ņØä ĒåĄĒĢ┤ MOG-immunoglobulin G (MOG-IgG)ļź╝ Ļ▓ĆņČ£ĒĢśļŖö Ļ▓āņØä ĻČīņןĒĢ£ļŗż. MOG-IgGĻ░Ć IgG1ņØ┤ĻĖ░ ļĢīļ¼ĖņŚÉ, Ļ▓Ćņé¼ņŚÉ ņō░ņØ┤ļŖö 2ņ░©ĒĢŁņ▓┤ļŖö IgG Fcļéś IgG1ļź╝ ņé¼ņÜ®ĒĢśļÉś, Ļ▓Ćņ”ØļÉ£ Ļ▓Ćņé¼ņŗżņØ╝ Ļ▓ĮņÜ░ IgG(H+L)ļź╝ ņé¼ņÜ®ĒĢĀ ņłś ņ׳ļŗż.39 Ļ│ĀņĀĢņäĖĒżĻĖ░ļ░śļČäņäØ(fixed cell-based assay)ņØĆ ņāØņäĖĒżĻĖ░ļ░śļČäņäØņØ┤ ļČłĻ░ĆļŖźĒĢĀ Ļ▓ĮņÜ░ ļīĆņ▓┤ Ļ▓Ćņé¼ļ▓Ģņ£╝ļĪ£ ņé¼ņÜ®ĒĢĀ ņłś ņ׳ņ¦Ćļ¦ī, ņāØņäĖĒżĻĖ░ļ░śļČäņäØļ▓Ģļ│┤ļŗż ļ»╝Ļ░ÉļÅäņÖĆ ĒŖ╣ņØ┤ļÅäĻ░Ć ļé«ņ£╝ļŗł ņŻ╝ņØśĒĢ┤ņĢ╝ ĒĢ£ļŗż. ĒÜ©ņåīĻ▓░ĒĢ®ļ®┤ņŚŁ ĒØĪņ░®ņĖĪņĀĢļ▓Ģ(enzyme-linked immunosorbent assay)ņØĆ ļ»╝Ļ░ÉļÅäņÖĆ ĒŖ╣ņØ┤ļÅäĻ░Ć ļé«ņĢä MOG-IgG ņĖĪņĀĢņŚÉļŖö ĻČīņןļÉśņ¦Ć ņĢŖļŖöļŗż.40
ŌĆśMOGĒĢŁņ▓┤ ļ¬ģĒÖĢĒĢ£ ņ¢æņä▒ŌĆÖņØś ĻĖ░ņżĆņØĆ ņāØņäĖĒżĻĖ░ļ░śļČäņäØņØ╝ Ļ▓ĮņÜ░ ņ¢æņä▒ĻĖ░ņżĆņŚŁĻ░Ćļ│┤ļŗż ņĄ£ņåī 2ļ▓ł ļŹö ĒؼņäØĒĢśņŚ¼ļÅä ņ¢æņä▒ņØ╝ ļĢīļéś ļČäņäØļ▓ĢņŚÉņä£ ņĀĢĒĢ£ ĒŖ╣ņĀĢ ņŚŁĻ░Ćļź╝ ļäśņØä ļĢī, ļśÉļŖö ĒśĢĻ┤æĒÖ£ņä▒ņäĖĒżņäĀļ│äĻĖ░(fluorescence-activated cell sorting)ļź╝ ņØ┤ņÜ®ĒĢ£ ņ£ĀņäĖĒżļČäņäØļ▓Ģ(flow cytometry) ĻĖ░ņżĆļ╣ä(cut-off ratio)ļź╝ ļäśņØä ļĢīļĪ£ ĒĢśļ®░, Ļ│ĀņĀĢņäĖĒżĻĖ░ļ░śļČäņäØņØ╝ Ļ▓ĮņÜ░ļŖö ņŚŁĻ░ĆĻ░Ć 1:100 ņØ┤ņāüņØ╝ ļĢīļĪ£ ĒĢ£ļŗż. ŌĆśMOG ĒĢŁņ▓┤ ņĢĮņ¢æņä▒ŌĆÖņØĆ ņāØņäĖĒżĻĖ░ļ░śļČäņäØņØĆ Ļ░ü ļČäņäØņŚÉņä£ ņĀĢĒĢ£ ņŚŁĻ░Ćļź╝ ĻĖ░ņżĆļ│┤ļŗż ļé«ņØä ļĢī, Ļ│ĀņĀĢņäĖĒżĻĖ░ļ░śļČäņäØņØĆ 1:10 ņØ┤ņāüņØ┤Ļ│Ā 1:100 ļ»Ėļ¦īņØ╝ ļĢīļĪ£ ĒĢ£ļŗż. MOGĒĢŁņ▓┤ņØś ņŚŁĻ░ĆĻ░Ć ļåÆņØĆ Ļ▓ĮņÜ░ ņŗżĒŚśņŗż Ļ░ä ņ×¼Ēśäņä▒ņØ┤ ņóŗņ¦Ćļ¦ī, ļé«ņØĆ Ļ▓ĮņÜ░ņŚÉļŖö ņŗżĒŚśņŗż Ļ░ä ņ¢æņä▒ ĒīÉļŗ©ņŚÉ ņ×¼Ēśäņä▒ņØ┤ ļ¢©ņ¢┤ņ¦äļŗż. ļśÉĒĢ£ ļåÆņØĆ ņŚŁĻ░Ćļź╝ ņĀüņÜ®ĒĢĀņłśļĪØ MOGĒĢŁņ▓┤Ļ▓Ćņé¼ņØś ņ¢æņä▒ņśłņĖĪļÅäĻ░Ć ļåÆņĢäņ¦äļŗż.40,41
ļŗ©ļÅģņ£╝ļĪ£ ļćīņ▓ÖņłśņĢĪņŚÉņä£ MOGĒĢŁņ▓┤ļź╝ Ļ▓Ćņé¼ĒĢśļ®┤ Ēśłņ▓Ł MOGĒĢŁņ▓┤Ļ▓Ćņé¼ļ│┤ļŗż ļ»╝Ļ░ÉļÅäĻ░Ć ļ¢©ņ¢┤ņ¦Ćņ¦Ćļ¦ī, ņØ╝ļČĆ Ēāłņłśņ┤łņ¦łĒÖś ĒÖśņ×ÉņŚÉņä£ Ēśłņ▓Ł MOGĒĢŁņ▓┤Ļ░Ć ņØīņä▒ņØ┤ļŹöļØ╝ļÅä ļćīņ▓ÖņłśņĢĪņŚÉņä£ļŖö MOGĒĢŁņ▓┤Ļ░Ć ņ¢æņä▒ņØ┤ņŚłĻ│Ā, ņØ┤ļōżņØ┤ MOGAD ņ×äņāü ņ¢æņāüņŚÉ ļČĆĒĢ®ĒĢśņśĆĻĖ░ ļĢīļ¼ĖņŚÉ, ņ×äņāüņĀü, ņśüņāüņØśĒĢÖņĀüņ£╝ļĪ£ MOGADĻ░Ć ņØśņŗ¼ļÉśļŖö Ļ▓ĮņÜ░ Ēśłņ▓Ł MOGĒĢŁņ▓┤Ļ░Ć ņØīņä▒ņØ┤ļØ╝ļ®┤, ļćīņ▓ÖņłśņĢĪ MOGĒĢŁņ▓┤Ļ▓Ćņé¼Ļ░Ć ļÅäņøĆņØ┤ ļÉĀ ņłś ņ׳ļŗż.42-44
Ļ▓Ćņé¼ ņŗ£ņĀÉņØĆ ņ×äņāü ņ”ØņāüņØ┤ ļéśĒāĆļé¼ņØä ļĢī, ĒŖ╣Ē׳ ĻĖēņä▒ĻĖ░ ņ╣śļŻī ņĀäņŚÉ Ļ▓Ćņé¼ĒĢĀ Ļ▓ĮņÜ░ Ļ░Ćņן MOGĒĢŁņ▓┤Ļ░Ć Ļ▓ĆņČ£ļÉĀ Ļ░ĆļŖźņä▒ņØ┤ ļåÆļŗż. ļ¦īņĢĮ ĻĖēņä▒ĻĖ░ ņ╣śļŻīĻ░Ć ņŗ£ņ×æļÉ£ ņØ┤Ēøä Ļ▓Ćņé¼Ļ░Ć ņŗ£Ē¢ēļÉśņŚłĻ│Ā, MOGĒĢŁņ▓┤Ļ░Ć ņØīņä▒ņØĖ Ļ▓ĮņÜ░, MOGADĻ░Ć ņØśņŗ¼ļÉ£ļŗżļ®┤, ĻĖēņä▒ĻĖ░ ņ╣śļŻī ĒÜ©Ļ│╝ ņóģļŻī Ēøä ņĄ£ņåī 3Ļ░£ņøöņØ┤ ņ¦Ćļé£ ļŗżņØīņØ┤ļéś, ņ×¼ļ░£ĒĢśņśĆņØä Ļ▓ĮņÜ░ ļŗżņŗ£ Ļ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢĀ Ļ▓āņØ┤ ĻČīņןļÉ£ļŗż.1,45
MOGAD ņ¦äļŗ© ĻĖ░ņżĆ
2023ļģä ņ▓śņØīņ£╝ļĪ£ MOGADņØś ņ¦äļŗ© ĻĖ░ņżĆņØ┤ ņĀ£ņĢłļÉśņŚłļŗż.3 MOGADņØś ņ¦äļŗ©ņØä ņ£äĒĢ┤ņä£ļŖö ņ×äņāü ņ¢æņāüĻ│╝ ņ”Øņāü ļ░£ņāØņØä ĒÖĢņØĖĒĢśĻĖ░ ņ£äĒĢ£ ņŗĀĻ▓ĮĒĢÖņĀü Ļ▓Ćņ¦äĻ│╝ ņśüņāüņØśĒĢÖņĀü ņåīĻ▓¼ ļ░Å Ļ▓Ćņé¼ņŗż ņåīĻ▓¼ņØä ļ¦īņĪ▒ĒĢ┤ņĢ╝ ĒĢ£ļŗż. ĒŖ╣Ē׳ ļ¬©ļōĀ Ēāłņłśņ┤łņ¦łĒÖśņŚÉņä£ ņŖżĒü¼ļ”¼ļŗØ ļ¬®ņĀüņ£╝ļĪ£ MOGĒĢŁņ▓┤Ļ▓Ćņé¼ļź╝ ĒĢśļŖö Ļ▓āņØĆ ņ¦Ćņ¢æĒĢ┤ņĢ╝ ĒĢ£ļŗż.3
ņĀ£ņŗ£ļÉ£ ņ¦äļŗ© ĻĖ░ņżĆņØĆ Table 2ņÖĆ Ļ░Öļŗż. ĒĢĄņŗ¼ņ×äņāüņ”Øņāüņ£ĀĒśĢ(core clinical attack type)Ļ│╝ Ēśłņ▓Ł ŌĆśMOGĒĢŁņ▓┤ ļ¬ģĒÖĢĒĢ£ ņ¢æņä▒ŌĆÖņØĖ Ļ▓ĮņÜ░, MOGADļĪ£ ņ¦äļŗ©ĒĢĀ ņłś ņ׳ļŗż. Ēśłņ▓Ł ŌĆśMOGĒĢŁņ▓┤ ņĢĮņ¢æņä▒ŌĆÖņØĖ Ļ▓ĮņÜ░, ņŚŁĻ░ĆĻ░Ć ņĀ£ņŗ£ļÉśņ¦Ć ņĢŖņØĆ Ļ│ĀņĀĢņäĖĒżĻĖ░ļ░śļČäņäØ ņ¢æņä▒ņØĖ Ļ▓ĮņÜ░, ļśÉļŖö Ēśłņ▓Ł ņØīņä▒ņØ┤ņ¦Ćļ¦ī ļćīņ▓ÖņłśņĢĪ ŌĆśMOGĒĢŁņ▓┤ ļ¬ģĒÖĢĒĢ£ ņ¢æņä▒ŌĆÖņØĖ Ļ▓ĮņÜ░ņŚÉļŖö ĒĢĄņŗ¼ņ×äņāüņ”Øņāüņ£ĀĒśĢņØ┤ ņ׳ļŹöļØ╝ļÅä ļ│┤ņĪ░ņ×äņāü/MRI ņ¢æņāü(supporting clinical or MRI features) ņżæ ĒĢ£ Ļ░£ ņØ┤ņāüņØ┤ MOGADļĪ£ ņ¦äļŗ©ĒĢĀ ņłś ņ׳ļŗż. ļ¬©ļōĀ ņ¦äļŗ©ņŚÉ ņĢ×ņä£ ļŹö ĒÖĢņŗżĒĢ£ ļŗżļźĖ ņ¦äļŗ©ņØ┤ ņ׳ļŗżļ®┤ ļ░░ņĀ£ļÉśņ¢┤ņĢ╝ ĒĢ£ļŗż. ĒŖ╣Ē׳ MSņØś Ļ▓ĮņÜ░ 0.3-2.5%ņŚÉņä£ MOGĒĢŁņ▓┤ ņ¢æņä▒ņØ┤ ļéśĒāĆļéĀ ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ņØ┤ļōżņØ┤ MOGADļĪ£ ņśżņ¦äļÉśņ¦Ć ņĢŖļÅäļĪØ ņŻ╝ņØśĒĢ┤ņĢ╝ ĒĢ£ļŗż. ļŗżļ¦ī, 11ņäĖ ļ»Ėļ¦īņØś ņåīņĢäņŚÉņä£ļŖö ņśżĒ׳ļĀż ņżæņČöņŗĀĻ▓ĮĻ│ä Ēāłņłśņ┤łņ¦łĒÖś ņżæņŚÉļŖö MSĻ░Ć ļō£ļ¼╝Ļ│Ā MOGĒĢŁņ▓┤ ņ¢æņä▒ņØĖ ĒÖśņĢäĻ░Ć ļ¦ÄĻĖ░ ļĢīļ¼ĖņŚÉ MOGĒĢŁņ▓┤Ļ▓Ćņé¼Ļ░Ć ĒĢäņÜöĒĢĀ ņłś ņ׳ļŗż. ĒĢŁņĢäņ┐ĀņĢäĒżļ”░4ĒĢŁņ▓┤ņÖĆ MOGĒĢŁņ▓┤Ļ░Ć ļæś ļŗż ņ¢æņä▒ņØĖ Ļ▓ĮņÜ░ļŖö ļ¦żņÜ░ ļō£ļ¼╝Ļ▓ī ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż. ĒĢśņ¦Ćļ¦ī ŌĆśMOGĒĢŁņ▓┤ ņĢĮņ¢æņä▒ŌĆÖņØ┤Ļ│Ā ĒĢŁņĢäņ┐ĀņĢäĒżļ”░4ĒĢŁņ▓┤Ļ░Ć ņ¢æņä▒ņØ┤ļØ╝ļ®┤ ņ¦äļŗ© ĻĖ░ņżĆņŚÉņä£ļÅä ņĀ£ņŗ£ļÉ£ ļ░öņÖĆ Ļ░ÖņØ┤ AQP4+NMOSD Ļ░ĆļŖźņä▒ņØ┤ ļŹö ļåÆļŗż. Ļ░Éļ│äņŚÉ ņŻ╝ņØśĒĢ┤ņĢ╝ĒĢśļŖö red-flag ņ”ØĒøäļĪ£ļŖö ŌæĀ ņ×äņāüņĀü ņ×¼ļ░£ ņŚåņØ┤ ņŗĀĻ▓ĮĒĢÖņĀü ņ”ØņāüņØś ņ¦ĆņåŹņĀü ņ¦äĒ¢ēĒĢśļŖö Ļ▓ĮņÜ░, ŌæĪ ņ”ØņāüņØś Ļ░Ćņן ņŗ¼ĒĢ£ ņĀĢņĀÉĻ╣īņ¦Ć ņłśļČäņŚÉņä£ ņłśņŗ£Ļ░ä ņĢłņŚÉ ņ┤łĻĖēņä▒ņ£╝ļĪ£ ļ░£ļ│æĒĢśļŖö Ļ▓ĮņÜ░, Ōæó ĻĖēņä▒ĻĖ░ ņŗ£ Ļ│ĀņÜ®ļ¤ē ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻīņŚÉ ĒśĖņĀäņØ┤ ņŚåļŖö Ļ▓ĮņÜ░, ŌæŻ ļŗżļ░£ņä▒Ļ▓ĮĒÖöņ”ØņŚÉ ņĀäĒśĢņĀüņØĖ MRI ņåīĻ▓¼Ļ│╝ ĒĢ©Ļ╗ś ņś¼ļ”¼Ļ│ĀĒü┤ļĪĀ ļØĀ ņ¢æņä▒ņØĖ Ļ▓ĮņÜ░, Ōæż ļ¼┤ņ”Øņāü T2 Ļ│ĀņŗĀĒśĖĻ░ĢļÅä ļ│æļ│ĆņØ┤ ļ░£ņāØĒĢśĻ▒░ļéś ĻĖ░ņĪ┤ ļ│æļ│ĆņØ┤ ļīĆļČĆļČä ĻĘĖļīĆļĪ£ ņ£Āņ¦ĆļÉśļŖö Ļ▓ĮņÜ░, Ōæź 6Ļ░£ņøö ņØ┤ņāü ņ¦ĆņåŹļÉśļŖö ņĪ░ņśüņ”ØĻ░Ģ ļ│æļ│ĆņØ┤ ņ׳ļŖö Ļ▓ĮņÜ░ ļō▒ņØ┤ ņ׳ļŗż.3
MOGADņØś ņ╣śļŻī
MOGADņØś ņ╣śļŻīļŖö ļŗżļźĖ ņżæņČöņŗĀĻ▓ĮĻ│ä ņŚ╝ņ”Ø Ēāłņłśņ┤łņ¦łĒÖśĻ│╝ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ĻĖēņä▒ĻĖ░ ņ╣śļŻīņÖĆ ņןĻĖ░ņĀü ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīļĪ£ ļéśļēĀ ņłś ņ׳ļŗż. ņŚ¼ĻĖ░ņä£ ņ×¼ļ░£ņØś ņĀĢņØśļŖö ņ▓½ ņ”ØņāüņØ┤ ļ░£ņāØĒĢ£ņ¦Ć 30ņØ╝ņØ┤ Ļ▓ĮĻ│╝ĒĢ£ Ēøä ņāłļĪ£ņÜ┤ ņ”ØņāüņØ┤ ļŗżņŗ£ ļéśĒāĆļé£ Ļ▓āņ£╝ļĪ£ ĒĢśņśĆļŗż.3
ĻĖēņä▒ĻĖ░ ņ╣śļŻī
Ēśäņ×¼Ļ╣īņ¦ĆļŖö MOGAD ĻĖēņä▒ĻĖ░ ņ╣śļŻīņŚÉ ļīĆĒĢ£ ļ¼┤ņ×æņ£ä ļīĆņĪ░ ņŗ£ĒŚś(randomized controlled trial)ņØ┤ļéś ĻĘ╝Ļ▒░ņżæņŗ¼(evidence-based) ņ¦Ćņ╣©ņØ┤ ņŚåļŗż. ļśÉĒĢ£ MOGĒĢŁņ▓┤ ņ¢æņä▒ ņŚ¼ļČĆĻ░Ć ņżæņČöņŗĀĻ▓ĮĻ│ä Ēāłņłśņ┤łņ¦łĒÖśņØś ĻĖēņä▒ĻĖ░ ņ╣śļŻīņŚÉ ņśüĒ¢źņØä ņżĆļŗżļŖö ĻĘ╝Ļ▒░Ļ░Ć ņŚåĻĖ░ ļĢīļ¼ĖņŚÉ, ļīĆļČĆļČä ņżæņČöņŗĀĻ▓ĮĻ│ä ņŚ╝ņ”Ø Ēāłņłśņ┤łņ¦łĒÖśņØś ĻĖēņä▒ĻĖ░ ņ╣śļŻīņŚÉ ļö░ļź┤Ļ│Ā ņ׳ļŗż. ĒŖ╣Ē׳, MOGĒĢŁņ▓┤Ļ▓Ćņé¼ Ļ▓░Ļ│╝ ĒÖĢņØĖņØ┤ ĻĖēņä▒ĻĖ░ ņ╣śļŻīļź╝ ņ¦ĆņŚ░ņŗ£ņ╝£ņä£ļŖö ņĢłļÉ£ļŗż.
ĻĖēņä▒ĻĖ░ ņ╣śļŻī 1ņ░© ņĢĮņĀ£ļŖö Ļ▓ĮņĀĢļ¦ź ļ®öĒŗĖĒöäļĀłļō£ļŗłņåöļĪĀņ£╝ļĪ£, 30 mg/kg/day ļśÉļŖö 1 g/day ņÜ®ļ¤ēņ£╝ļĪ£ 3-5ņØ╝ ņŚ░ņåŹņ£╝ļĪ£ ņ╣śļŻīĒĢ£ļŗż. MOGADļŖö ņŚ¼ļ¤¼ Ļ┤Ćņ░░ ņŚ░ĻĄ¼ņŚÉņä£ Ļ▓ĮņĀĢļ¦ź Ļ│ĀņÜ®ļ¤ē ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻī ļ░śņØæņØ┤ ņóŗņØĆ Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż.23,46 ņ╣śļŻī ĒÜ©Ļ│╝Ļ░Ć ņČ®ļČäņ╣ś ņĢŖņØĆ Ļ▓ĮņÜ░, ĒśłņןļČäļ”¼ĻĄÉĒÖśņłĀ(plasmapheresis), ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░ ņĀĢļ¦ź ņŻ╝ņé¼(intravenous immunoglobulin, IVIG), ļśÉļŖö ĒśłņןļČäļ”¼ĻĄÉĒÖśņłĀ Ēøä IVIGļź╝ Ļ│ĀļĀżĒĢĀ ņłś ņ׳ļŗż.47 ĒśłņןļČäļ”¼ĻĄÉĒÖśņłĀņØĆ ĒåĄņāüņĀüņ£╝ļĪ£ļŖö ļ¦żļ▓ł Ēśłņן ļČĆĒö╝ņØś 1-1.5ļ░░ ņĀĢļÅäļź╝ ĻĄÉņ▓┤ĒĢśļ®░, Ļ▓®ņØ╝ļĪ£ ņ┤Ø 5-7ļ▓ł ņŗ£Ē¢ēĒĢ£ļŗż. ļō£ļ¼╝Ļ▓ī ņżæņŗ¼ņĀĢļ¦źĻ┤Ć ņéĮņ×ģĻ│╝ Ļ┤ĆļĀ©ļÉ£ ņČ£ĒśłņØ┤ļéś Ļ░ÉņŚ╝ ņ£äĒŚś, ņŗ£ĒŖĖļź┤ņé░ņŚÉ ņØśĒĢ£ ņĀĆņ╣╝ņŖśĒśłņ”Ø, ļīĆņé¼ņé░ņ”Ø ļō▒ņØ┤ ļ░£ņāØĒĢĀ ņłś ņ׳ļŗż. ĒŖ╣Ē׳, Ēśłņן ņĢäļŗī ņĢīļČĆļ»╝ ļō▒ņ£╝ļĪ£ ĻĄÉņ▓┤ĒĢĀ Ļ▓ĮņÜ░ ņØæĻ│ĀņØĖņ×ÉĻ░Ć Ļ▓░ĒĢŹļÉĀ ņłś ņ׳ņ£╝ļ»ĆļĪ£ ņŻ╝ņØśĒĢ┤ņĢ╝ ĒĢ£ļŗż. IVIGļŖö ņØ╝ļ░śņĀüņ£╝ļĪ£ 0.4 g/kg/day ņÜ®ļ¤ēņØä 5ņØ╝ ļÅÖņĢł Ēł¼ņŚ¼ĒĢśņŚ¼ ņ┤Ø 2 g/kgņØä Ēł¼ņŚ¼ĒĢ£ļŗż. ļČĆņ×æņÜ®ņØĆ ļō£ļ¼╝Ļ▓ī ļæÉĒåĄ, ļ¼┤ļĀźĻ░É, ņŚ┤, ļ░£ņ¦ä, ĒśłņĢĪņØæĻ│ĀņØ┤ņāü, ņŗĀņןĻĖ░ļŖźņĀĆĒĢś, ņĢäļéśĒĢäļØĮņŗ£ņŖż ļō▒ņØ┤ ļ░£ņāØĒĢĀ ņłś ņ׳ļŗż.
Ļ│ĀņÜ®ļ¤ē Ļ▓ĮņĀĢļ¦ź ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻī ĒøäņŚÉ Ļ▓ĮĻĄ¼ņÜ® ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻī ņŚ¼ļČĆ, ĻĖ░Ļ░ä, ņÜ®ļ¤ēņØĆ ņ”ØņāüņØś ņżæņ”ØļÅä, ņ×¼ņĢģĒÖö ņ£äĒŚśņä▒ņØä Ļ│ĀļĀżĒĢśņŚ¼ Ļ▓░ņĀĢĒĢśļŖöļŹ░, MOGADļŖö ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻīņŚÉ ņØśņĪ┤ņä▒ņØ┤ ļåÆņĢä Ļ░Éļ¤ē ļśÉļŖö ņżæļŗ© ņŗ£ņŚÉ ņ×¼ļ░£ĒĢĀ ņ£äĒŚśņä▒ņØ┤ ņ׳Ļ│Ā, ņ×¼ļ░£ ļśÉĒĢ£ 2Ļ░£ņøö ņØ┤ļé┤ņŚÉ ļ¦ÄņØ┤ ļ░£ņāØĒĢśļ»ĆļĪ£, ņĄ£ņåī 3Ļ░£ņøö ņØ┤ņāüņŚÉ Ļ▒Ėņ│É ņä£ņä£Ē׳ Ļ░Éļ¤ēĒĢśļŖö Ļ▓āņØ┤ ĻČīņןļÉ£ļŗż.24
ņןĻĖ░ņĀü ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻī
ņżæņČöņŗĀĻ▓ĮĻ│ä ņŚ╝ņ”Ø Ēāłņłśņ┤łņ¦łĒÖśņŚÉņä£ ņ×¼ļ░£ņä▒ Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖö ņ¦łĒÖśņØś Ļ▓ĮņÜ░ ņןĻĖ░ņĀüņØĖ ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢśņ¦Ćļ¦ī, MOGADļŖö ļŗ©ļ░£ņä▒ Ļ▓ĮĻ│╝ņÖĆ ņ×¼ļ░£ņä▒ Ļ▓ĮĻ│╝Ļ░Ć ļ¬©ļæÉ Ļ░ĆļŖźĒĢ£ ņ¦łĒÖśņØ┤ļŗż. ņĢäņ¦üņØĆ MOGAD ĒÖśņ×ÉņŚÉņä£ ņ×¼ļ░£ĒĢĀ ņ£äĒŚśņä▒ņØä ĒÅēĻ░ĆĒĢśĻ▒░ļéś ņןĻĖ░ņĀü ņśłĒøäļź╝ ņśłņĖĪĒĢĀ ņłś ņ׳ļŖö ļ¬ģĒÖĢĒĢ£ ņśłņĖĪņØĖņ×ÉļŖö ņŚåļŗż. ĒŖ╣Ē׳ ņåīņĢäņŚÉņä£ļŖö ņĢĮ 70% ņĀĢļÅäņØś ĒÖśņ×ÉĻ░Ć ļŗ©ļ░£ņä▒ ņ”ØņāüņŚÉ ĻĘĖņ╣śĻĖ░ ļĢīļ¼ĖņŚÉ, ņןĻĖ░ņĀüņØĖ ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīļź╝ ņŗ£ņ×æĒĢ┤ņĢ╝ ĒĢĀņ¦Ć Ļ▓░ņĀĢĒĢśĻĖ░ļŖö ļŹöņÜ▒ ņ¢┤ļĀĄļŗż. ļŗżļ¦ī ņ×¼ļ░£ņØ┤ ņ׳ļŖö ĒÖśņ×ÉņŚÉņä£ļŖö ņןĻĖ░ņĀüņØĖ ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīļź╝ ņŗ£ņ×æĒĢĀ Ļ▓āņØ┤ ĻČīņןļÉ£ļŗż. ĻĘĖ ņÖĖņŚÉ ņ╣śļŻī ņŚ¼ļČĆļź╝ Ļ▓░ņĀĢĒĢśļŖö ļŹ░ņŚÉļŖö ņ▓½ ņ”Øņāü ņ╣śļŻīņŗ£ ņ╣śļŻī ļ░śņØæ, ņ▓½ ņ”ØņāüņØś ņżæņ”ØļÅä, ņןņĢĀ ņČĢņĀü ņ£äĒŚśņä▒, ņןĻĖ░ņĀü ļ®┤ņŚŁ ņ╣śļŻīņØś ņ£äĒŚśņä▒ ļō▒ņØä Ļ│ĀļĀżĒĢśņŚ¼ņĢ╝ ĒĢ£ļŗż.24 ņśłļź╝ ļōżņ¢┤, ņ▓½ ņ”Øņāü Ēøä ņןņĢĀ ņĀĢļÅäĻ░Ć ņŗ¼ĒĢśņŚ¼ ņĀüĻĘ╣ņĀüņ£╝ļĪ£ ņ×¼ļ░£ņØä ņśłļ░®ĒĢ┤ņĢ╝ ĒĢśļŖö Ļ▓ĮņÜ░ļéś ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻīņŚÉ ļ¦żņÜ░ ņØśņĪ┤ņĀüņØĖ ņ”Øņāü Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░ļŖö ņ▓½ ļ░£ļ│æņØ┤ļŹöļØ╝ļÅä ņןĻĖ░ņĀü ļ®┤ņŚŁņ¢ĄņĀ£ ņ╣śļŻīļź╝ Ļ│ĀļĀżĒĢ┤ļ│╝ ņłś ņ׳Ļ▓Āļŗż. MOG ĒĢŁņ▓┤ņØś ņØīņĀä ņŚ¼ļČĆ ļśÉĒĢ£ ņ×¼ļ░£Ļ│╝ Ļ┤ĆļĀ©ņØ┤ ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ņŻ╝ĻĖ░ņĀüņ£╝ļĪ£ MOGĒĢŁņ▓┤Ļ▓Ćņé¼ļź╝ ĒĢśņŚ¼ ļ®┤ņŚŁ ņ╣śļŻī ņŚ¼ļČĆļź╝ Ļ▓░ņĀĢĒĢśļŖö ļŹ░ņŚÉ ņ░ĖĻ│ĀĒĢĀ ņłś ņ׳Ļ▓Āļŗż.
ņןĻĖ░ ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīņŚÉ ļīĆĒĢ┤ņä£ Ēśäņ×¼ ļ¼┤ņ×æņ£ä ļīĆņĪ░ĻĄ░ ņ×äņāü ņŚ░ĻĄ¼Ļ░Ć ņ¦äĒ¢ēļÉśĻ│Ā ņ׳ņ£╝ļéś ņĢäņ¦ü ņÖäļŻīļÉ£ ņŚ░ĻĄ¼Ļ░Ć ņŚåĻĖ░ ļĢīļ¼ĖņŚÉ, ĒøäĒ¢źņĀü ņŚ░ĻĄ¼, Ļ┤Ćņ░░ ņŚ░ĻĄ¼, ņŗżņĀ£ņ×äņāüņ×ÉļŻī(real world data)ļź╝ ļ░öĒāĢņ£╝ļĪ£ ņĢĮļ¼╝ņØä ņé┤ĒÄ┤ļ│┤Ļ│Āņ×É ĒĢ£ļŗż. ņĢäņ×ÉĒŗ░ņśżĒöäļ”░(azathioprine, 2-3 mg/kg/day), ļ»ĖņĮöĒÄśļåĆļĀłņØ┤ĒŖĖļ¬©ĒÄśĒŗĖ(mycophenolate mofetil, 2,000 mg/day)Ļ│╝ Ļ░ÖņØĆ Ļ▓ĮĻĄ¼ņÜ® ļ®┤ņŚŁņ¢ĄņĀ£ņĀ£, ļ”¼ĒłŁņŗ£ļ¦Ö Ļ░ÖņØĆ BņäĖĒż ņ¢ĄņĀ£ņĀ£, ņŻ╝ĻĖ░ņĀü ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░ ņĀĢņŻ╝(IVIG) ļō▒ņØ┤ ņŚ░Ļ░ä ņ×¼ļ░£ļźĀ(annualized relapse rate)ņØä ļé«ņČöļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĪīļŗż. ļŗżļ¦ī MSņŚÉņä£ ņé¼ņÜ®ĒĢśļŖö ņ¦łļ│æņĪ░ņĀłņĢĮņĀ£(disease modifying drug)ļŖö MOGAD ņ×¼ļ░£ ļ░®ņ¦ĆņŚÉ ĒÜ©Ļ│╝Ļ░Ć ņŚåņŚłļŗż. ņĢäņ×ÉĒŗ░ņśżĒöäļ”░, ļ»ĖņĮöĒÄśļåĆļĀłņØ┤ĒŖĖļ¬©ĒÄśĒŗĖĻ│╝ Ļ░ÖņØĆ Ļ▓ĮĻĄ¼ņÜ® ļ®┤ņŚŁņ¢ĄņĀ£ņĀ£ļź╝ ņé¼ņÜ®ĒĢ£ ĒÖśņ×ÉņŚÉņä£ļŖö Ļ░üĻ░ü ņĢĮ 39%, 47% ĒÖśņ×ÉņŚÉņä£ ņ×¼ļ░£ņØä ļ¦ēņØä ņłś ņ׳ņŚłļŗż.18,23,24,48-51
ļ”¼ĒłŁņŗ£ļ¦ÖņØä ņé¼ņÜ®ĒĢ£ ĒÖśņ×ÉņŚÉņä£ļŖö ņĢĮ 50%ņŚÉņä£ ņ×¼ļ░£ņØä ļ¦ēņØä ņłś ņ׳ņŚłļŖöļŹ░, ņØ┤ļŖö AQP4+NMOSDņŚÉņä£ņØś ņ╣śļŻī ņä▒ņĀüĻ│╝ ļ╣äĻĄÉĒĢ┤ņä£ļŖö ĒÜ©Ļ│╝Ļ░Ć ņĪ░ĻĖł ņĀüņØĆ Ļ▓āņØä ņĢī ņłś ņ׳ļŗż.52 ļ”¼ĒłŁņŗ£ļ¦Ö(rituximab)ņØĆ CD20ņØä ĒāĆĻ╣āņ£╝ļĪ£ ĒĢśļŖö BņäĖĒż ņ¢ĄņĀ£ņĀ£ļĪ£, ņŗ£ņ×æņÜöļ▓ĢņØĆ 1) 375 mg/m2 ņÜ®ļ¤ēņ£╝ļĪ£ 1ņŻ╝ļ¦łļŗż 4ņŻ╝ ļÅÖņĢł Ēł¼ņĢĮĒĢśĻ▒░ļéś, 2) 1 gņØä 2ņŻ╝ļ¦łļŗż 2ĒÜī Ēł¼ņĢĮĒĢśļŖö ļ░®ļ▓ĢņØ┤ Ļ░ĆļŖźĒĢśĻ│Ā, ņØ┤Ēøä ņ£Āņ¦Ć ņ╣śļŻīļŖö 375 mg/m2 ļśÉļŖö 1 gņØä CD19ļź╝ ļ¬©ļŗłĒä░ĒĢśņŚ¼ 1% ņ┤łĻ│╝ ņŗ£ņŚÉ ļśÉļŖö 6Ļ░£ņøöļ¦łļŗż Ēł¼ņŚ¼ĒĢ£ļŗż.
ņŻ╝ĻĖ░ņĀü IVIGļŖö ņĢĮ 69%ņØś ĒÖśņ×ÉņŚÉņä£ ņ×¼ļ░£ņØä ļ¦ēņĢśņ£╝ļ®░, ņØ╝ļČĆ ĒÖśņ×ÉļōżņŚÉņä£ļŖö EDSS ņĀÉņłśņØś ĒśĖņĀä ļśÉĒĢ£ ļ│┤Ļ│ĀļÉśņŚłļŗż. ņØ╝ļ░śņĀüņ£╝ļĪ£ 4ņŻ╝ Ļ░äĻ▓®ņ£╝ļĪ£ 0.4-2 g/kgņØś ņÜ®ļ¤ēņ£╝ļĪ£ ņŻ╝ĻĖ░ņĀü Ēł¼ņŚ¼ĒĢĀ ņłś ņ׳ļŗż. ļ¬ćļ¬ć ĒøäĒ¢źņĀü ņŚ░ĻĄ¼ņŚÉņä£ ņ×¼ļ░£ ļ░®ņ¦Ć ĒÜ©Ļ│╝Ļ░Ć ļŗżļźĖ ļ®┤ņŚŁņ¢ĄņĀ£ņĀ£ļ│┤ļŗż ņóŗļŗżļŖö ņØśĻ▓¼ņØ┤ ņ׳ņ£╝ļéś, ņĄ£ļīĆ 50%ņØś ĒÖśņ×ÉņŚÉņä£ ņ×¼ļ░£ņØ┤ ļ│┤Ļ│ĀļÉśņŚłĻ│Ā, ĒŖ╣Ē׳ ļé«ņØĆ ņÜ®ļ¤ēņØ╝ ļĢī ļśÉļŖö Ēł¼ņŚ¼ Ļ░äĻ▓®ņØ┤ ņŚ░ņןļÉĀ ļĢī ņ×¼ļ░£ņØś ņ£äĒŚśņä▒ņØ┤ ļåÆņØĆ Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłļŗż.
ĻĘĖ ņÖĖņŚÉļÅä ĒåĀņŗżļ”¼ņŻ╝ļ¦Ö(tocilizumab)ņØĆ ņØĖĒä░ļŻ©Ēé©-6 ņłśņÜ®ņ▓┤ ņ░©ļŗ©ņĀ£ļĪ£ ņåīĻĘ£ļ¬© ņŚ░ĻĄ¼ņŚÉņä£ ņĢĮ73% ĒÖśņ×ÉņŚÉņä£ ņ×¼ļ░£ņØä ļ¦ēņĢśņ£╝ļ®░, ļŗżļźĖ ļ®┤ņŚŁņ¢ĄņĀ£ ņ╣śļŻīņŚÉ ļČłņØæĒĢśļŖö ĒÖśņ×ÉņŚÉņä£ 8 mg/kgņ£╝ļĪ£ ņøö 1ĒÜī Ēł¼ņŚ¼(ņĄ£ļīĆ 800 mg/ņøö)ļź╝ ņŗ£ļÅäĒĢ┤ļ│╝ ņłś ņ׳Ļ▓Āļŗż.53 ĒĢśņ¦Ćļ¦ī ņĢäņ¦ü ņןĻĖ░ ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīļź╝ ĒĢ┤ņĢ╝ ĒĢĀ ĒÖśņ×ÉņØś ņäĀļ│ä, ņ╣śļŻī ĻĖ░Ļ░ä, ņ╣śļŻī ņżæļŗ©ņŚÉ ļīĆĒĢ£ ļŹö ļ¦ÄņØĆ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśļŗż.
MOGADņØś ņ×äņāü Ļ▓ĮĻ│╝ ļ░Å ņśłĒøä
ņ×äņāüņĀü ņ×¼ļ░£ņØĆ ņ▓½ ņ”Øņāü ļ░£ņāØ Ēøä 6Ļ░£ņøö ņØ┤ļé┤Ļ░Ć ņØ┤Ēøäļ│┤ļŗż ļŹö ņ×”ļŗż. ĒŖ╣Ē׳ Ļ▓ĮĻĄ¼ ņŖżĒģīļĪ£ņØ┤ļō£ ņ╣śļŻīļź╝ ņżäņØ┤Ļ▒░ļéś ņżæļŗ©ĒĢ£ ņØ┤Ēøä 2Ļ░£ņøö ņØ┤ļé┤ņŚÉ ņ×ÉņŻ╝ ņ×¼ļ░£ĒĢ£ļŗż.23,46 ņä▒ņØĖĻ│╝ ņåīņĢäņŚÉņä£ ņ×¼ļ░£ņØś ļ╣łļÅäņŚÉ ņ░©ņØ┤Ļ░Ć ņ׳ļŖöļŹ░, ņåīņĢäņŚÉņä£ļŖö ņ×¼ļ░£ņØ┤ ņĀüĻ│Ā ļŗ©ļ░£ĒśĢ Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░Ļ░Ć ļ¦Äļŗż.19,54 ņŚ¼ļ¤¼ ņןĻĖ░ ņČöņĀü ņĮöĒśĖĒŖĖņŚÉņä£ 5ļģä ņØ┤ļé┤ ņ×¼ļ░£ļźĀņØĆ ņåīņĢäņŚÉņä£ ņĢĮ 17-30%,47,54,55 ņä▒ņØĖņŚÉņä£ļŖö ņĢĮ 40-62% ņĀĢļÅäļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż.42,46,56 ļō£ļ¼╝ņ¦Ćļ¦ī MOGAD ĒÖśņ×ÉņŚÉņä£ ĒŖ╣Ē׳ ņ▓½ ņ”Øņāü ļ░£ņāØ Ēøä 1ļģä ņØ┤ļé┤ņŚÉ ļ¼┤ņ”Øņāü ļćī ļ│æļ│ĆņØ┤ ņČöĻ░ĆļĪ£ ņāØĻĖĖ ņłś ņ׳ļŗż. ļŗżļ¦ī ņØ┤ļ¤¼ĒĢ£ ļ¼┤ņ”Øņāü ļćī ļ│æļ│ĆņØ┤ ņāØĻĖ░ļŖö Ļ▓ĮņÜ░ Ē¢źĒøä ņ×¼ļ░£ĒĢĀ ņ£äĒŚśņä▒ņØ┤ ļåÆņØīņØä ņŗ£ņé¼ĒĢĀ ņłś ņ׳ļŗż.57 MSņÖĆļŖö ļŗ¼ļ”¼ ņ×äņāüņĀü ņ×¼ļ░£ ņŚåņØ┤ ņ¦łļ│æņØ┤ ņ¦äĒ¢ēĒĢśļŖö Ļ▓āņØĆ ļ¦żņÜ░ ļō£ļ¼╝ņ¦Ćļ¦ī ņČöĻ░ĆņĀüņØĖ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśļŗż.
ņ▓śņØī ņ”Øņāü ļ░£ņāØ ļŗ╣ņŗ£ MOGĒĢŁņ▓┤ņØś ņŚŁĻ░ĆļŖö ĒÜīļ│ĄņØ┤ļéś ņ×¼ļ░£ņØä ņśłņĖĪĒĢĀ ņłś ņŚåļŗż. ļ░śļ│Ą ņ▒äĒśłļĪ£ MOGĒĢŁņ▓┤ļź╝ Ļ▓Ćņé¼ĒĢĀ Ļ▓ĮņÜ░ ņ¦ĆņåŹņĀüņØĖ ĒĢŁņ▓┤ ņ¢æņä▒, ĒŖ╣Ē׳ ļåÆņØĆ ņŚŁĻ░ĆņØś MOGĒĢŁņ▓┤ļź╝ Ļ░Ćņ¦ä Ļ▓ĮņÜ░ ĒĢŁņ▓┤Ļ░Ć ņØīņĀäļÉśļŖö Ļ▓ĮņÜ░ļ│┤ļŗż ļåÆņØĆ ņ×¼ļ░£ļźĀĻ│╝ ņŚ░Ļ┤ĆļÉśņ¢┤ ņ׳ņŚłļŗż.
ņåīņĢäņŚÉņä£ļŖö ļīĆņ▓┤ļĪ£ ļŗ©ļ░£ņä▒ Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░Ļ░Ć ļ¦ÄĻ│Ā, EDSS 3ņĀÉņŚÉ ļÅäļŗ¼ĒĢśļŖö ņÜ┤ļÅÖņןņĢĀĻ░Ć ļ░£ņāØĒĢśļŖö ļ╣äņ£© ļśÉĒĢ£ 10% ļ»Ėļ¦ī(ņä▒ņØĖņŚÉņä£ļŖö 20-40%)ņ£╝ļĪ£ ņśłĒøäĻ░Ć ņóŗņØĆ ĒÄĖņØ┤ļéś, ņØ╝ļČĆ ĒÖśņ×ÉņŚÉņä£ ļ░▒ņ¦łĒśĢņä▒ņןņĢĀ ņ£Āņé¼ ļ│æļ│Ćņ£╝ļĪ£ ļ░£ĒśäĒĢśļŖö Ļ▓ĮņÜ░ ņśłĒøäĻ░Ć ņóŗņ¦Ć ņĢŖņ£╝ļŗł ņŻ╝ņØśĒĢ┤ņĢ╝ ĒĢ£ļŗż. MSņÖĆ ļŗ¼ļ”¼ ņ¦äĒ¢ēņä▒ Ļ▓ĮĻ│╝ļŖö ļ¦żņÜ░ ļō£ļ¼╝Ļ│Ā, MSļéś AQP4+NMOSD ļ¦īĒü╝ ņ×¼ļ░£ņä▒ Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░Ļ░Ć ļ¦Äņ¦ĆļŖö ņĢŖņ¦Ćļ¦ī ņØ╝ļČĆ ĒÖśņ×ÉņŚÉņä£ļŖö ļ╣łļ▓łĒĢ£ ņ×¼ļ░£Ļ│╝ ļ░śļ│ĄņĀüņØĖ ņןņĢĀņØś ņČĢņĀüņ£╝ļĪ£ ļČłļ¤ēĒĢ£ ņśłĒøäļź╝ ļ│┤ņŚ¼ņŻ╝ļŖö Ļ▓ĮņÜ░Ļ░Ć ņ׳ņ¢┤ ņĀüĻĘ╣ņĀüņØĖ ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢśļŗż.
Ļ▓░ļĪĀ
MOGĒĢŁņ▓┤ņØś ņĀĢĒÖĢĒĢ£ Ļ▓ĆņČ£ ļ░®ļ▓ĢņØ┤ ņĀ£ņŗ£ļÉśļ®┤ņä£ļČĆĒä░ MOGADļØ╝ļŖö ņāłļĪ£ņÜ┤ ņ¦äļŗ© ļ▓öņŻ╝Ļ░Ć ņāØĻĖ░Ļ│Ā, ņØ┤ ļČäņĢ╝ņŚÉ ĒÖ£ļ░£ĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ņØ┤ļŻ©ņ¢┤ņ¦ĆĻ│Ā ņ׳ļŗż. ļ│æĒā£ņāØļ”¼ļź╝ ļ░ØĒ׳Ļ│Ā, ļ░£ļ│æ ĻĖ░ņĀäņØä ņØ┤ĒĢ┤ĒĢśļŖö Ļ▓āņØĆ ļ│æņØś Ļ▓ĮĻ│╝ļź╝ ĒīīņĢģĒĢśļŖö ļ░öņØ┤ņśżļ¦łņ╗ż ļ░£ĻĄ┤ ļ░Å ņ╣śļŻīņĀ£ Ļ░£ļ░£ņŚÉļÅä ņżæņÜöĒĢśļŗż. MOGADļŖö ņ×äņāü ņ¢æņāüņØ┤ ļ¦żņÜ░ ļŗżņ¢æĒĢśĻ│Ā, ņĀäĒśĢņĀüņØĖ MOGADĻ░Ć ņĢäļŗī ĒÖśņ×Éļéś Ļ▒┤Ļ░ĢĒĢ£ ņé¼ļ×īņŚÉņä£ļÅä MOGĒĢŁņ▓┤Ļ░Ć ņ¢æņä▒ņØ╝ ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ, MOGAD Ēæ£ĒśäĒśĢņŚÉ ņĀüĒĢ®ĒĢ£ ĒÖśņ×ÉņŚÉņä£ ņĀĢĒÖĢĒĢ£ ļ░®ļ▓Ģņ£╝ļĪ£ ĒĢŁņ▓┤Ļ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢśĻ│Ā ĻĘĖ Ļ▓░Ļ│╝ļź╝ ĒĢ┤ņäØĒĢśļŖö Ļ▓āņØ┤ ņ£äņ¢æņä▒ ļ░Å ņśżņ¦äņØä ņżäņØ┤Ļ│Ā Ļ▓Ćņé¼ņØś ņ¢æņä▒ņśłņĖĪļÅäļź╝ ļåÆņØ┤ļŖö ļŹ░ņŚÉ ņżæņÜöĒĢśļŗż. ņĢäņ¦ü ņ╣śļŻī ĒÜ©Ļ│╝ļź╝ ņĀĢĒÖĢĒĢśĻ▓ī ĒīÉļŗ©ĒĢĀ ņłś ņ׳ļŖö ņŚ░ĻĄ¼Ļ░Ć ņŚåņ¢┤, Ēśäņ×¼ ņ¦äĒ¢ēļÉśĻ│Ā ņ׳ļŖö ļ¼┤ņ×æņ£ä ļīĆņĪ░ĻĄ░ ņ×äņāüņŗ£ĒŚśņØś Ļ▓░Ļ│╝ ļśÉĒĢ£ ĻĖ░ļīĆĻ░Ć ļÉ£ļŗż. ļŗżļ¦ī MSļéś AQP4+NMOSDņÖĆ Ļ░ÖņØ┤ ļ¬©ļōĀ ĒÖśņ×ÉĻ░Ć ņןĻĖ░ ņ×¼ļ░£ ļ░®ņ¦Ć ņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢ£ Ļ▓āņØĆ ņĢäļŗłĻĖ░ ļĢīļ¼ĖņŚÉ ņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢ£ ĒÖśņ×Éļź╝ ņĪ░ĻĖ░ņŚÉ ņäĀļ│äĒĢĀ ņłś ņ׳ļŖö ļ░®ļ▓ĢņØ┤ ĒĢäņÜöĒĢśļŗż.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print