Myelin-oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is an inflammatory demyelinating disorder of the central nervous system (CNS) mediated by antibodies, and has been recognized as a novel inflammatory disease entity independent of neuromyelitis optica spectrum disorders (NMOSDs) and multiple sclerosis (MS) over the last few years.1-3 Patients with MOGAD may develop any combination of optic neuritis (ON), myelitis, brainstem syndrome, encephalitis, or acute disseminated encephalomyelitis (ADEM), which is estimated to have an annual incidence of 1.6/million person years.4-7 MOG, a molecule found on the external membrane of myelin sheaths, was shown to regulate the stability of oligodendrocyte microtubules, maintain the structural integrity of the myelin sheath, and more importantly, function as an essential target of autoantibodies and cell-mediated immune responses in MOGAD.8-10 MOG is thought to exist mainly within the CNS including the optic nerves. Consequently, the clinical phenotype of MOGAD includes combinations described above and fulminant disease progression.10
Among post-infective CNS diseases, ADEM usually develops following an acute viral etiology, especially exanthematous disease, bacterial infection, or vaccination but rarely after immune sera.11 There are two proposed explanations for the development of ADEM. The first is the inflammatory cascade concept, which involves direct invasion of the neurotropic pathogens into the CNS. The second is molecular mimicry between the pathogen and myelin proteins of the host.11
Therefore, in patients with inflammatory demyelinating lesions of the CNS after a preceding infection, differentiation between ADEM and MOGAD is often challenging to diagnose, and the pathogenesis of CNS demyelination induced by infection remains unclear.
Herein, we report a case of MOGAD secondary to upper respiratory infection in a young woman presenting with ADEM.
CASE
A 27-year-old woman, with no relevant medical history, presented with rapidly progressing weakness in all four extremities and decreased mental function with accompanying headache, vomiting, and urinary difficulty after upper respiratory infection for a week and was admitted to our department. She reported no past medical history of immune-related or neurological diseases, and there was no familial history of immune-related disease. One week before the onset of quadriparesis, she had a mild cough and fever that resolved spontaneously without treatment. On presentation at our neurologic department, neurological examination revealed drowsy mentality, dysarthria, dysphagia, increased deep tendon reflex, positive Babinski sign, HoffmanŌĆÖs sign, and decreased motor power with Medical Research Council grade 2 strength in the upper and lower extremities. Pinprick, temperature, vibration, and proprioception sensation were decreased from the C4 spinal cord level. Hoffman's and Tromner's sign were increased in both hands.
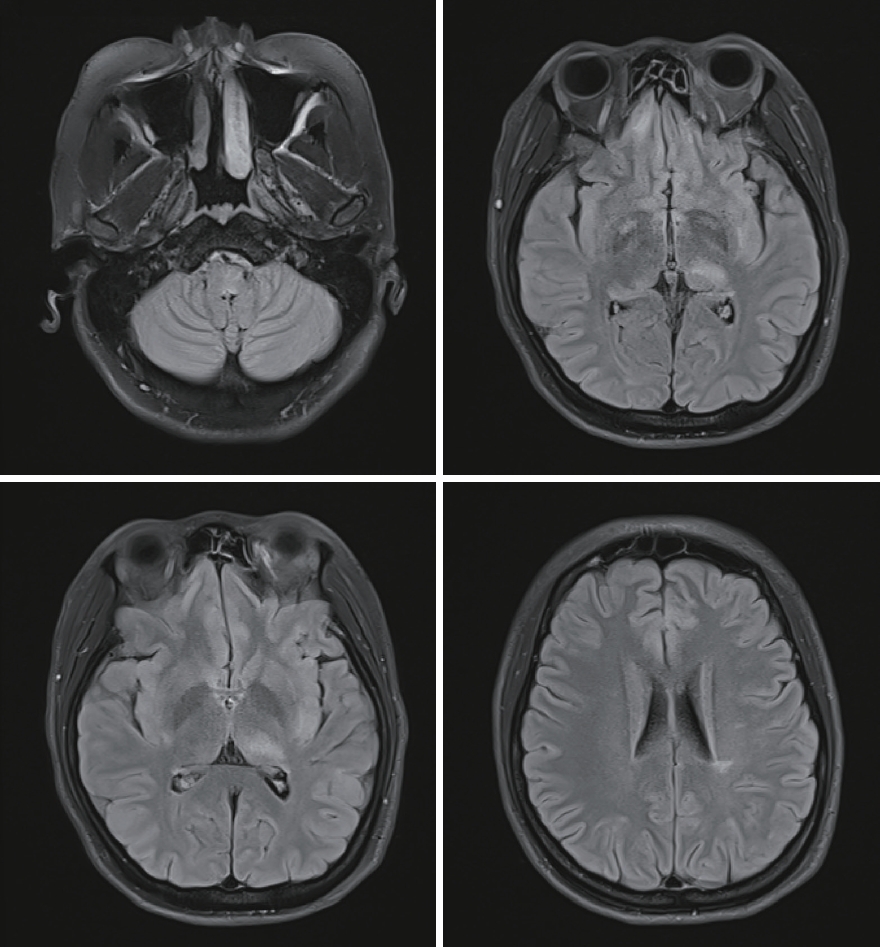
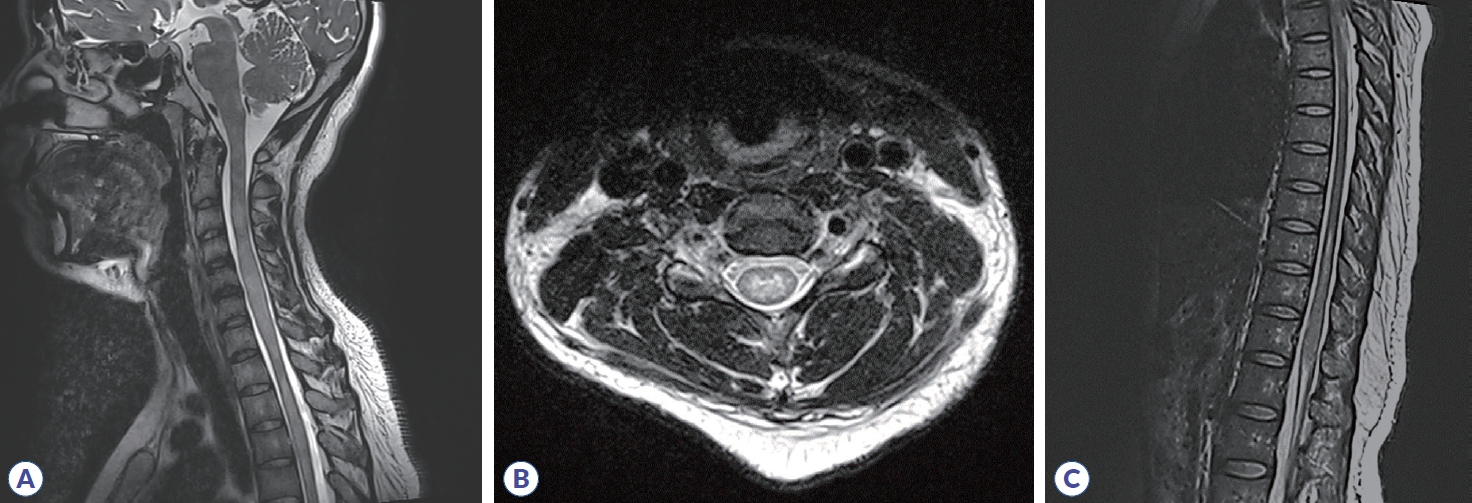
Initial brain MRI using fluid-attenuated inversion recovery revealed inflammatory lesions in the left corona radiata/centrum semiovale, left insula, right basal ganglia, right postcentral gyrus, and left dorsal medulla (Fig. 1). In addition, spine MRI revealed increased T2 signal from the 3rd to 7th segment of the cervical spine, the 6th to 12th segment of the thoracic spine, and the medullary cone had abnormal hyperintensity with edematous expansion of the cord and weak enhancement (Fig. 2). CSF analysis revealed an increase in white blood cells (40 mm3, lymphocyte dominant) and protein (141.2 mg/dL) without oligoclonal band. She was initially considered to have infectious encephalomyelitis or ADEM. Due to the possibility of immune-related inflammatory brain diseases including MS, NMOSD, autoimmune encephalitis and vasculitis, specific laboratory tests were performed and autoimmune antibodies evaluated, targeting neuronal cell surface antigens, ion channels (voltage-gated potassium channels), ligand-gated ion channels (N-methyl-D-aspartate, ╬▒-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, and gamma-aminobutyric acid-b receptor channels). In addition, she was tested for rheumatoid factor, lupus anticoagulant, anti-SSA/SSB, and antiaquaporin-4 antibodies; specific abnormalities were not found. Conclusively, elevated serum MOG-IgG antibodies were detected using a clinically validated fluorescence-activated cell sorting (FACS) live cell assay and she was diagnosed with MOGAD involving brain and spinal cord. The ratio of positive cells (RPCs) was 0.937 (positive >0.254) and mean fluorescence intensity ratio (MFIr) was 16.82 (positive >3.62).
Initially, the patient was treated for 5 consecutive days with high-dose intravenous methylprednisolone (1 g/day), followed by high-dose oral prednisolone (60 mg/day). After treatment, the mental state and muscle power of the patient improved and symptoms such as headache and vomiting gradually resolved. After treatment with high-dose steroid therapy, oral prednisone with mycophenolate was given as maintenance therapy.
Two months after discharge, MRI was performed and the initial abnormal signals in the brain and spinal cord were significantly decreased; however, serum MOG-IgG remained positive. Follow-up MOG titers were 0.836 (RPCs) and 19.06 (MFIr). Since then, the patient has continued maintenance immunosuppressive treatment with oral prednisolone and mycophenolate.
DISCUSSION
MOGAD is a group of inflammatory demyelinating diseases of the CNS. According to the diagnostic criteria proposed by Banwell et al.12 in 2023, patients with one of the following core clinical attack types and positive serum MOG-IgG test results measured using fixed or live cell-based assay can be diagnosed with MOGAD: a) core clinical demyelinating event (ON/myelitis/ADEM/cerebral monofocal or polyfocal deficits/brainstem or cerebellar deficits/cerebral cortical encephalitis often with seizures); b) positive MOG-IgG test with supporting clinical or MRI features. The patient had the clinical manifestations of quadriparesis, hyperalgesia, and urinary retention. MRI of the head showed multiple demyelinating lesions in the brain and MRI of the spine showed long-segment myelopathy involving the cervical, thoracic, and conus cords.5-9 Combined with the positive results of serum MOG-IgG, the diagnosis of MOGAD was confirmed. ADEM usually occurs as a result of early infection or vaccination. MOGAD presenting as ADEM secondary to respiratory infection has been rarely reported and the pathogenesis of preceding infection causing MOGAD remains unclear. However, we believe this is a case of MOGAD secondary to preceding infection, which has potential value in elucidating the pathophysiology of MOGAD.
Several pathophysiological mechanisms of infection-related CNS inflammatory disorders have been proposed, including direct viral neuroinvasion, cytokine-induced CNS inflammation, hypercoagulability, and/or hypoxia.4 MOG-IgG antibodies are postulated to cause demyelination through various mechanisms, including antibody-dependent cytotoxicity and encephalitogenic T-cells.
In conclusion, after an upper respiratory infection, our patient showed symptoms of ADEM with accompanying headache, vomiting, lethargy, and urinary difficulty, and she was diagnosed with MOGAD based on FACS live cell assay. High-dose steroids and an immunosuppressant as well as proper management of the MOGAD patient have markedly improved the neurological symptoms and MRI abnormalities.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print